
Iodine chemistry in the coastal marine atmosphere
ECB Bulletin July 2009
The sources, abundance and impacts of halogen compounds have been widely studied in atmospheric chemistry for several decades, with much attention previously focussed upon the involvement of man-made chlorine and bromine species in stratospheric ozone depletion. In this article, ECG committee members William Bloss and Stephen Ball consider a different angle on atmospheric halogen chemistry, looking at the role of naturally occurring iodine species in the marine boundary layer (the lowermost layer of the atmosphere over the oceans). The focus is on mid-latitude coastal regions, where a number of recent field measurements have elucidated aspects of iodine’s sources, its behaviour and the impacts of its chemistry on the atmospheric environment.Iodine sources and atmospheric chemistry in the marine environment
The primary sources of iodine species to the marine atmosphere are iodocarbons (RI) such as CH2I2, CH2IBr and CH3I, and molecular iodine, I2 [see for example the reviews by Carpenter (2003) and von Glasow & Crutzen (2007)]. Iodocarbons are released by various marine organisms, including macroalgae (seaweeds), which live in coastal regions, and phytoplankton living in the surface waters of the ocean. Atmospheric mixing ratios of iodocarbons are typically around the pptv level, although they can be significantly higher in regions of high biological productivity (pptv = parts per trillion by volume: 1 part in 1012). The major source of I2 is seaweeds, particularly species of kelps such as Laminaria digitata which can have an iodine content as high as 1 % of their dry mass (Verhaeghe et al., 2008). As we will see below, in coastal regions I2 release from seaweeds can be overwhelming and can dominate other iodine sources.
The biological drivers for halogen release remain unclear. Studies on seaweeds indicate that iodine species are released as a stress response to factors such as desiccation and exposure to UV light or ozone. Iodine may also be involved in the physiological protection of the plant from toxic active oxygen species such as hydrogen peroxide. Haloperoxidase enzymes are thought to catalyse the reaction of hydrogen peroxide with halide anions, leading to the formation of hypoiodous acid, HOI (Palmer et al., 2005). Subsequent production of iodocarbons may occur via the haloform reaction, which involves the substitution of hydrogen atoms adjacent to a carbonyl group for halogen atoms, either within the plant cells, or in organic matter suspended in seawater. It is also possible that iodine is produced in seawater by inorganic reactions involving dissolved ozone, iodide (I-(aq)) and HOI.
Once released to the atmosphere, molecular iodine and most iodocarbons undergo solar photolysis on timescales ranging from seconds (I2) to minutes (e.g. CH2I2) to a few days (e.g. CH3I), leading to the release of constituent iodine atoms into the lower atmosphere.
Once released to the atmosphere, molecular iodine and most iodocarbons undergo solar photolysis on timescales ranging from seconds (I2) to minutes (e.g. CH2I2) to a few days (e.g. CH3I), leading to the release of constituent iodine atoms into the lower atmosphere.
I2, RI + hv → I
It is worth noting that the timescales for liberating free iodine atoms are very much faster than the timescales for the release of chlorine or bromine atoms from long-lived anthropogenic chlorofluorocarbons which only undergo significant photolysis in the stratosphere (typical lifetime = tens of years). Thus iodine compounds simply don’t persist long enough in the atmosphere to be mixed upwards and affect stratospheric ozone chemistry. However, iodine atoms do react with ozone close to the surface, leading to the formation of the gas-phase IO radical which lies at the centre of marine boundary layer iodine chemistry (Figure 1). Subsequent reactions of IO can regenerate iodine atoms leading to catalytic ozone destruction cycles, for example via some channels of the IO self-reaction:
2 x I + O3 → IO + O2
IO + IO → I + I + O2
Net 2 O3 → 3 O2
(All reactions refer to gas-phase species unless otherwise indicated)
IO + IO → I + I + O2
Net 2 O3 → 3 O2
(All reactions refer to gas-phase species unless otherwise indicated)
|
Ozone acts as a greenhouse gas in the troposphere, and is a pollutant harmful to human health, vegetation and certain materials, for example some plastics and rubber. Consequently, processes that affect ozone concentrations are of considerable interest to atmospheric chemists. Tropospheric ozone also acts as the precursor to the key atmospheric oxidant, the hydroxyl radical (OH). The OH radical acts as a natural cleansing agent for the atmosphere. The reaction with OH initiates (and is usually the rate determining step in) the degradation of many organic compounds, pollutants and greenhouse gases such as methane emitted into the atmosphere from both natural and anthropogenic sources. Processes which affect ozone and/or OH abundance can therefore affect atmospheric oxidising capacity.
|
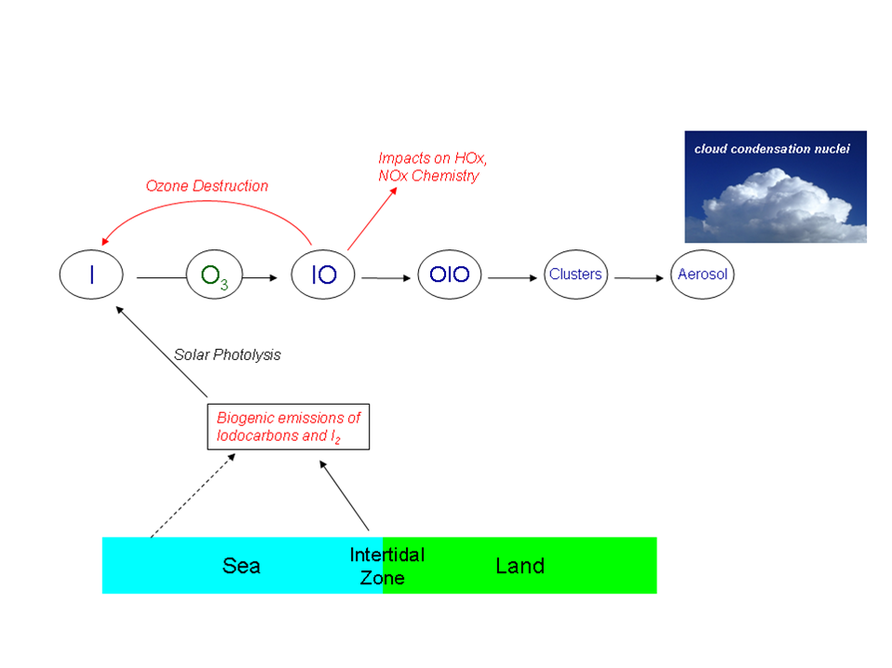
Figure 1: The atmospheric chemistry of inorganic iodine species in the marine boundary layer
|
The IO radical also interacts with other atmospheric radicals, including HOx (OH + HO2), for example via the sequence:
IO + HO2 → HOI + O2
HOI + hv → OH + I
I + O3 → IO + O 2
Net HO2 + O3 + hv → OH + 2O2
HOI + hv → OH + I
I + O3 → IO + O 2
Net HO2 + O3 + hv → OH + 2O2
The formation and photolysis of HOI therefore converts HO2 to OH, again affecting atmospheric oxidising capacity. A further effect of this reaction follows from the heterogeneous loss of HOI (i.e. removal of HOI from the gas phase to aerosol particles). This leads to a reduction in OH and HO2 levels, as HOx is removed from the gas-phase system. Concomitantly the halogens can be returned to the gas-phase, or even undergo an amplification in concentration, through acid-catalysed aqueous phase reactions within aerosol particles (below; X,Y = Cl / Br / I) which liberate the (lower solubility) di-halogen / inter-halogen compounds back into the gas phase:
HOx + aerosol → HOX (aq)
HOx (aq) + Y− (aq) + H+(aq) → H2O(l) + XY
HOx (aq) + Y− (aq) + H+(aq) → H2O(l) + XY
A further, and rather different, effect of iodine chemistry is to drive the formation of new particles in the atmosphere (or the growth of pre-existing ones). Laboratory studies of gas-phase iodine reactions have long been afflicted by the formation of white deposits upon optical surfaces when appreciable levels of iodine oxides are produced in the presence of ozone. Recently chamber experiments and atmospheric observations have shown that polymerisation reactions, thought to involve the combination of OIO monomers (formed through the self-reaction of IO radicals), can form clusters and ultimately solid-phase particles of bulk formula I2O5 and/or I4O9 (McFiggans et al., 2004; Saunders & Plane, 2005).
IO + IO → OIO + I
n OIO ( + m O3 ?) → → (I2O5)x (s)
→ → bulk phase iodate (IO3−)
n OIO ( + m O3 ?) → → (I2O5)x (s)
→ → bulk phase iodate (IO3−)
Hence the biogenic release of iodine source gases could amplify the number concentration and/or the size of marine aerosol particles. If an aerosol particle can then grow sufficiently by the condensation of ambient water vapour, it may act as a cloud condensation nucleus (CCN). Particles that contain a high fraction of soluble inorganic salts (here, iodate) are thought to be particularly efficient cloud condensation nuclei. Higher CCN concentrations lead to the formation of clouds composed of more, but smaller, water droplets which are brighter (reflecting more solar radiation) and which persist for longer. Thus, the contribution of iodine species to the marine particle population could have a significant climate influence (O’Dowd et al., 2002). Understanding the details and quantifying the magnitude of these effects are key issues, which recent laboratory and field measurements have begun to unravel.
Measurements of iodine chemistry
The atmospheric science community’s expanding interest in iodine chemistry in recent years is being driven both by a desire to understand some interesting science and, more practically, by substantial advances achieved in analytical instrumentation which now make it possible to detect iodine compounds at the very high dilutions found in the ambient atmosphere. For instance an automated GC-MS instrument has been developed for iodocarbons, with impressive detection limits down to around 0.05 pptv (Carpenter et al., 1999). Here atmospheric samples are drawn through an adsorbent trap to pre-concentrate the iodocarbons; the trap is then heated to drive off adsorbed molecules which are separated by gas chromatography and detected by mass spectrometry. A pre-concentration methodology (but with a different detector) has been used for denuder sampling atmospheric I2 into starch solutions (e.g. Saiz-Lopez et al., 2006). However, I2 and, conveniently, also its oxides IO and OIO absorb light at visible wavelengths, and their structured absorption spectra make them particularly amenable to in situ spectroscopic detection. Thus any of these three iodine compounds can be identified unambiguously in an atmospheric sample’s absorption spectrum via the molecule’s characteristic absorption bands and, provided the molecular absorption cross sections are known accurately from lab measurements, the absorption bands’ intensities provide a direct measure of ambient concentration (without the need for further calibrations). The disadvantage of deploying an absorption measurement for atmospheric sampling is that the low ambient concentrations of the target species means that observations need to be conducted over a very long absorption path through the atmosphere in order to produce a detectable signal. Long-path differential optical absorption spectroscopy (DOAS) achieves path lengths of ≈10 km by reflecting its light beam off a distant retro-reflector (a mirror). Indeed atmospheric IO and OIO were first observed by DOAS instruments (e.g. Alicke et al., 1999), and now such methods also routinely detect I2.
The atmospheric science community’s expanding interest in iodine chemistry in recent years is being driven both by a desire to understand some interesting science and, more practically, by substantial advances achieved in analytical instrumentation which now make it possible to detect iodine compounds at the very high dilutions found in the ambient atmosphere. For instance an automated GC-MS instrument has been developed for iodocarbons, with impressive detection limits down to around 0.05 pptv (Carpenter et al., 1999). Here atmospheric samples are drawn through an adsorbent trap to pre-concentrate the iodocarbons; the trap is then heated to drive off adsorbed molecules which are separated by gas chromatography and detected by mass spectrometry. A pre-concentration methodology (but with a different detector) has been used for denuder sampling atmospheric I2 into starch solutions (e.g. Saiz-Lopez et al., 2006). However, I2 and, conveniently, also its oxides IO and OIO absorb light at visible wavelengths, and their structured absorption spectra make them particularly amenable to in situ spectroscopic detection. Thus any of these three iodine compounds can be identified unambiguously in an atmospheric sample’s absorption spectrum via the molecule’s characteristic absorption bands and, provided the molecular absorption cross sections are known accurately from lab measurements, the absorption bands’ intensities provide a direct measure of ambient concentration (without the need for further calibrations). The disadvantage of deploying an absorption measurement for atmospheric sampling is that the low ambient concentrations of the target species means that observations need to be conducted over a very long absorption path through the atmosphere in order to produce a detectable signal. Long-path differential optical absorption spectroscopy (DOAS) achieves path lengths of ≈10 km by reflecting its light beam off a distant retro-reflector (a mirror). Indeed atmospheric IO and OIO were first observed by DOAS instruments (e.g. Alicke et al., 1999), and now such methods also routinely detect I2.
DOAS’s typical 5-km-out-5-km-back sampling geometry means that the concentrations inferred from such a measurement represent the average concentration of a given molecule over the entire light path. This presents a problem when looking at molecular iodine chemistry because I2 emissions from seaweeds tend to be localised on the shoreline, and because the I2 atmospheric lifetime is extremely short these emissions are not evenly mixed along the DOAS light path. A “point measurement” analogue of DOAS is provided by newly-developed cavity-based methods such as broadband cavity ringdown spectroscopy (Ball & Jones, 2003) and broadband cavity enhanced absorption spectroscopy (Langridge et al., 2008) which achieve multi-kilometre absorption paths by repeated reflections of a light beam inside a high finesse optical cavity composed of extremely efficient mirrors (> 99.99% reflection efficiency). Certain species can also be quantified by fluorescence spectroscopy, again as localised point measurements. Thus atmospheric IO has been detected using laser induced fluorescence (Whalley et al., 2007) using an instrument originally developed to detect the even-less abundant OH radical, and a resonance fluorescence instrument has been deployed to measure the ambient iodine atoms, with a trial deployment showing a strong correlation between iodine activity and daytime low tide (Bale et al., 2008).
A consistent observational picture is beginning to emerge from field studies, particular those conducted at costal sites with abundant seaweed populations (e.g. Mace Head, Galway, Ireland; Roscoff, Brittany, France). Generally the concentration of iodine compounds peaks around low tide when the seaweed beds are exposed to the atmosphere. IO and I atom concentrations peak sharply at daytime low tides because their source is atmospheric photochemistry; conversely, I2 concentrations peak at night-time low tides because during the day a substantial fraction of the emitted I2 is probably photolysed during its short wind-borne journey from seaweed source to the instruments located on dry land (see Figures 2 and 3). However, all three species are more abundant for the very lowest low tides, again suggesting that the source is from seaweed and particularly the deeper-water seaweeds that are exposed only rarely by unusually low tides. The more elusive OIO radical remains something of an enigma – some observations correlate with tide, but others do not. However, the rapid production of large numbers of very small iodine-rich aerosol particles in “nucleation bursts” do strongly correlate with low tides and their elevated I2 levels (Saiz-Lopez et al., 2006).
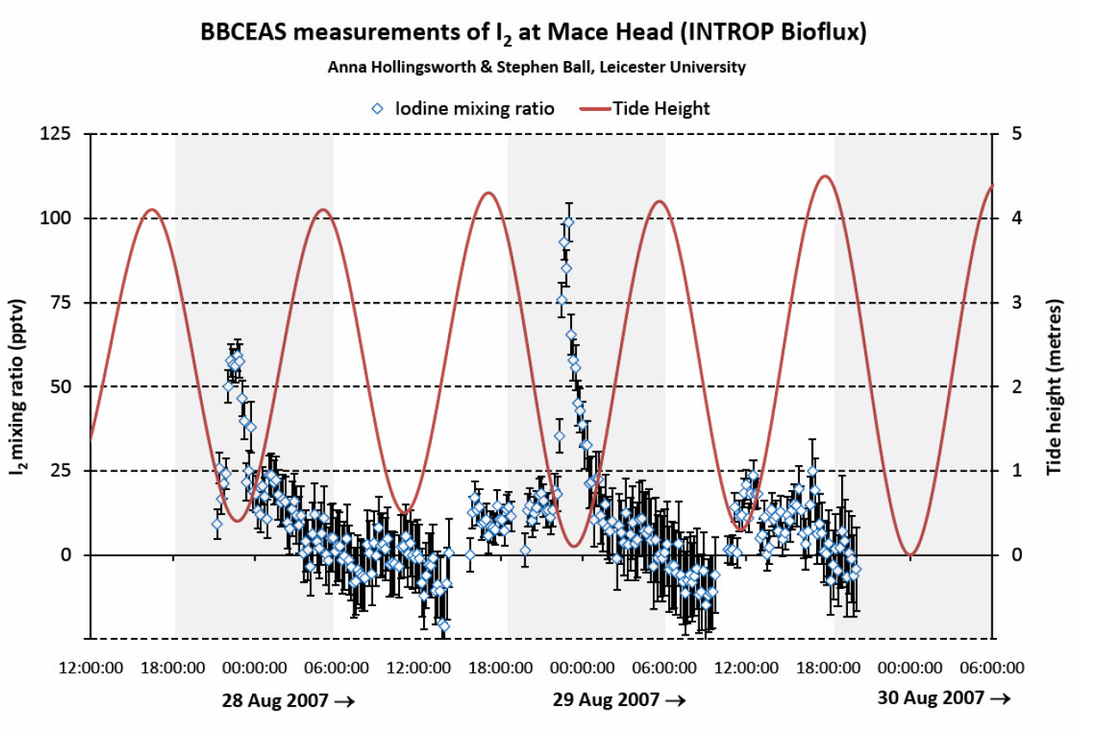
Figure 2: Molecular iodine, I2, measured on the shoreline at Mace Head, Ireland, using broadband cavity-enhanced spectroscopy (BBCEAS) (a photo of the instrument appears in Figure 4). I2 levels peak at low tide, at night (shaded regions)
|
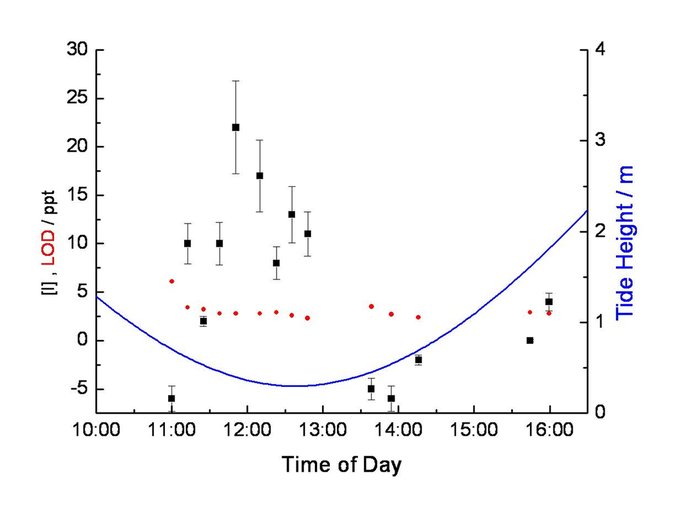
Figure 3: Iodine atoms measured on the shoreline at Mace Head, Ireland using resonance fluorescence detection (see also Figure 4 and Bale et al., 2008). Figure shows observed I mixing ratios (black points), detection limit (red circles) and local tide height (blue line; right hand axis). Iodine levels peak at low tide, at midday, when I2 emission and photolysis are both maximised
|
Outlook
While recent field, laboratory and theoretical studies have led to significant advances in our understanding of the role of iodine species in the troposphere, a number of major uncertainties remain.
The discussion above has focused upon observations at mid-latitude coastal sites, but these represent a small fraction of the global marine environment. Is iodine activity also found in the open ocean at significant levels? Recent observations from the Cape Verde observatory, located in the tropical mid-Atlantic remote from coastal influences, have measured IO at levels of 2 pptv (Read et al., 2008) with associated ozone destruction rates of 1.8 ppb day-1. The latter may be compared with a total (boundary layer) ozone level of 30 - 35 ppbv, i.e. the halogen-driven ozone destruction is significant (ppbv = parts per billion, by volume). Again these observations were only from a single location. However, the first satellite retrievals of IO (Saiz-Lopez et al., 2007; Schönhardt et al., 2008) have shown substantial iodine columns over the Southern Ocean (associated with sea ice coverage) and in lower latitude regions such as the Eastern Pacific. The satellite data cannot accurately determine the altitude distribution of the IO column density retrieved, so the concentrations at specific altitudes cannot be determined, and further ground-based field observations will be required to assess the global marine boundary layer impact of iodine chemistry.
While recent field, laboratory and theoretical studies have led to significant advances in our understanding of the role of iodine species in the troposphere, a number of major uncertainties remain.
The discussion above has focused upon observations at mid-latitude coastal sites, but these represent a small fraction of the global marine environment. Is iodine activity also found in the open ocean at significant levels? Recent observations from the Cape Verde observatory, located in the tropical mid-Atlantic remote from coastal influences, have measured IO at levels of 2 pptv (Read et al., 2008) with associated ozone destruction rates of 1.8 ppb day-1. The latter may be compared with a total (boundary layer) ozone level of 30 - 35 ppbv, i.e. the halogen-driven ozone destruction is significant (ppbv = parts per billion, by volume). Again these observations were only from a single location. However, the first satellite retrievals of IO (Saiz-Lopez et al., 2007; Schönhardt et al., 2008) have shown substantial iodine columns over the Southern Ocean (associated with sea ice coverage) and in lower latitude regions such as the Eastern Pacific. The satellite data cannot accurately determine the altitude distribution of the IO column density retrieved, so the concentrations at specific altitudes cannot be determined, and further ground-based field observations will be required to assess the global marine boundary layer impact of iodine chemistry.
A second issue is our understanding of the fundamental photochemical and kinetic parameters describing the behaviour of iodine species in the atmosphere. A number of these are uncertain or simply not known (for example, the absorption cross sections of various iodine reservoir compounds, and the mechanism and kinetics of the route from OIO through I2O4 and on to formation of clusters and hence new particles). These uncertainties hamper our ability to quantitatively model the impact of a given iodine loading upon the atmosphere, and will require further laboratory studies for their resolution.
Finally, the biological mechanism for the production of iodine species and their physiological function within the plant remains unclear – and consequently so does our understanding of any response the system might have to changing conditions, such as climate or ocean acidity, which may affect the marine biota ultimately responsible for much of the atmospheric iodine production. Addressing these and other questions is the subject of ongoing research activity within the atmospheric chemistry community, which will improve our understanding of the behaviour of this intriguing species in our atmosphere.

Figure 4: Novel instruments for in situ measurements of reactive iodine species deployed at the Mace Head site. Left panel: A dual channel broadband Cavity-Enhanced Absorption Spectrometer for observations of IO, OIO and I2 – photograph courtesy of Anna Hollingsworth.
|

Figure 4: Novel instruments for in situ measurements of reactive iodine species deployed at the Mace Head site. Right panel: Resonance Fluorescence instrument for the detection of iodine atoms and photolabile iodine species
|
References
Alicke, B.; Hebestreit, K.; Stutz, J.; Platt, U. (1999), Iodine oxide in the marine boundary layer, Nature, 397, 572.
Bale, C. S. E.; Ingham, T.; Commane, R.; Heard, D. E.; Bloss, W. J. (2008), Novel measurements of atmospheric iodine species by resonance fluorescence, J. Atmos. Chem., 60, 51.
Ball, S. M.; Jones R. L. (2003), Broadband cavity ringdown spectroscopy, Chem. Rev., 103, 5239.
Carpenter, L. J.; Sturges, W. T.; Liss, P. S.; Penkett, S. A.; Alicke B.; Hebestreit, K.; Platt, U. (1999), Short-lived alkyl-iodides and bromides at Mace Head: Links to macroalgal emission and halogen oxide formation, J. Geophys. Res., 104(D1), 1679.
Carpenter, L. J. (2003), Iodine in the marine boundary layer, Chem. Rev., 103, 4953.
Langridge, J. M.; Ball, S. M.; Shillings, A. J. L.; Jones, R. L. (2008), A broadband absorption spectrometer using light emitting diodes for ultrasensitive in situ trace gas detection, Rev. Sci. Instr., 79, 123110.
McFiggans, G. et al. (2004), Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine, Atmos. Chem. Phys., 4, 701.
O’Dowd, C. D.; Jimenez, J. L.; Bahreini, R.; Flagan, R. C.; Seinfeld, J. H.; Hämeri, K.; Pirjola, L.; Kulmala, M.; Jennings, S. G.; Hoffmann, T. (2002), Marine particle formation by biogenic iodine emissions, Nature, 417, 632.
Palmer, C. J.; Anders, T. L.; Carpenter, L. J.; Küpper, F. C.; McFiggans, G. B. (2005), Iodine and halocarbon response of Laminaria digitata to oxidative stress and links to atmospheric new particle production, Environ. Chem., 2, 282.
Read, K.A. et al. (2008), Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 453, 1232.
Saiz-Lopez, A.; Plane, J. M. C.; McFiggans, G.; Williams, P. I.; Ball, S. M.; Bitter, M.; Jones, R. L.; Hongwei, C.; Hoffmann, T. (2006), Modelling molecular iodine emissions in a coastal marine environment: the link to new particle formation, Atmos. Chem. Phys., 6, 883.
Saiz-Lopez, A.; Chance, K.; Liu, X.; Kurosu, T. P.; Sander, S. P. (2007), First observations of iodine oxide from space, Geophys. Res. Lett., 34, doi:10.1029/2007GL030111.
Saunders, R. W.; Plane, J. M. C. (2005), Formation pathways and composition of iodine oxide ultra-fine particles, Environ. Chem., 2, 299.
Schönhardt, A.; Richter, A.; Wittrock, F.; Kirk, H.; Oetjen, H.; Roscoe, H. K.; Burrows, J. P. (2008), Observations of iodine monoxide columns from satellite, Atmos. Chem. Phys., 8, 637.
Verhaeghe, E. F.; Fraysse, A.; Guerquin-Kern, J-C.; Wu, T.-D.; Deves, G.; Mioskowski, C.; LeBlanc, C.; Ortega, R.; Ambroise, Y.; Potin, P. (2008), Microchemical imaging of iodine distribution in the brown alga Laminaria digitata suggests a new mechanism for its accumulation, J. Biol. Inorg. Chem., 13, 257.
von Glasow, R.; Crutzen, P. J. (2007), Tropospheric halogen chemistry, In Treatise on Geochemistry, 4, 1.
Whalley, L. K.; Furneaux, K. L.; Gravestock, T.; Atkinson, H. M.; Bale, C. S. E.; Ingham, T.; Bloss, W. J.; Heard, D. E. (2007), Detection of iodine monoxide radicals in the marine boundary layer using laser induced fluorescence spectroscopy, J. Atmos. Chem., 58, 19.
Web link: The Mace Head Research Facility http://macehead.nuigalway.ie/mace1.html
Alicke, B.; Hebestreit, K.; Stutz, J.; Platt, U. (1999), Iodine oxide in the marine boundary layer, Nature, 397, 572.
Bale, C. S. E.; Ingham, T.; Commane, R.; Heard, D. E.; Bloss, W. J. (2008), Novel measurements of atmospheric iodine species by resonance fluorescence, J. Atmos. Chem., 60, 51.
Ball, S. M.; Jones R. L. (2003), Broadband cavity ringdown spectroscopy, Chem. Rev., 103, 5239.
Carpenter, L. J.; Sturges, W. T.; Liss, P. S.; Penkett, S. A.; Alicke B.; Hebestreit, K.; Platt, U. (1999), Short-lived alkyl-iodides and bromides at Mace Head: Links to macroalgal emission and halogen oxide formation, J. Geophys. Res., 104(D1), 1679.
Carpenter, L. J. (2003), Iodine in the marine boundary layer, Chem. Rev., 103, 4953.
Langridge, J. M.; Ball, S. M.; Shillings, A. J. L.; Jones, R. L. (2008), A broadband absorption spectrometer using light emitting diodes for ultrasensitive in situ trace gas detection, Rev. Sci. Instr., 79, 123110.
McFiggans, G. et al. (2004), Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine, Atmos. Chem. Phys., 4, 701.
O’Dowd, C. D.; Jimenez, J. L.; Bahreini, R.; Flagan, R. C.; Seinfeld, J. H.; Hämeri, K.; Pirjola, L.; Kulmala, M.; Jennings, S. G.; Hoffmann, T. (2002), Marine particle formation by biogenic iodine emissions, Nature, 417, 632.
Palmer, C. J.; Anders, T. L.; Carpenter, L. J.; Küpper, F. C.; McFiggans, G. B. (2005), Iodine and halocarbon response of Laminaria digitata to oxidative stress and links to atmospheric new particle production, Environ. Chem., 2, 282.
Read, K.A. et al. (2008), Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 453, 1232.
Saiz-Lopez, A.; Plane, J. M. C.; McFiggans, G.; Williams, P. I.; Ball, S. M.; Bitter, M.; Jones, R. L.; Hongwei, C.; Hoffmann, T. (2006), Modelling molecular iodine emissions in a coastal marine environment: the link to new particle formation, Atmos. Chem. Phys., 6, 883.
Saiz-Lopez, A.; Chance, K.; Liu, X.; Kurosu, T. P.; Sander, S. P. (2007), First observations of iodine oxide from space, Geophys. Res. Lett., 34, doi:10.1029/2007GL030111.
Saunders, R. W.; Plane, J. M. C. (2005), Formation pathways and composition of iodine oxide ultra-fine particles, Environ. Chem., 2, 299.
Schönhardt, A.; Richter, A.; Wittrock, F.; Kirk, H.; Oetjen, H.; Roscoe, H. K.; Burrows, J. P. (2008), Observations of iodine monoxide columns from satellite, Atmos. Chem. Phys., 8, 637.
Verhaeghe, E. F.; Fraysse, A.; Guerquin-Kern, J-C.; Wu, T.-D.; Deves, G.; Mioskowski, C.; LeBlanc, C.; Ortega, R.; Ambroise, Y.; Potin, P. (2008), Microchemical imaging of iodine distribution in the brown alga Laminaria digitata suggests a new mechanism for its accumulation, J. Biol. Inorg. Chem., 13, 257.
von Glasow, R.; Crutzen, P. J. (2007), Tropospheric halogen chemistry, In Treatise on Geochemistry, 4, 1.
Whalley, L. K.; Furneaux, K. L.; Gravestock, T.; Atkinson, H. M.; Bale, C. S. E.; Ingham, T.; Bloss, W. J.; Heard, D. E. (2007), Detection of iodine monoxide radicals in the marine boundary layer using laser induced fluorescence spectroscopy, J. Atmos. Chem., 58, 19.
Web link: The Mace Head Research Facility http://macehead.nuigalway.ie/mace1.html