
GEOSPEC abstracts Geochemical speciation & bioavailability of trace elements (GeoSpec2010)
ECG Bulletin January 2011
Six out of the 33 posters which featured at the GeoSpec2010 meeting held at Lancaster University last September have been expanded by their authors into papers for the ECG Bulletin. Extended extracts from these six papers appear below and the complete (partly edited) papers accompany the web version of the January 2011 ECG Bulletin (www.rsc.org/ecg and links).
Paper 1 Lessons from a large scale deployment of DGT in the Seine basin
Emmanuelle Uher1, Cécile Mirande-Bret2, Catherine Gourlay-Francé1
1. Cemagref , Parc de Tourvoie, 92163 Antony, France
2. LEESU, ENPC, Cité Descartes, 77455 Marne La Vallée, France
Introduction
The link between the chemical speciation of metals and their toxicity [1] has resulted in a number of different analytical techniques to assess the various fractions of metals present in the aqueous environment. Among these, the Diffusive Gradient in Thin-film technique (DGT), developed by Zhang and Davison [2], is commonly used. DGT is an in situ speciation method; it samples a labile fraction composed of free inorganic metals and weakly labile organic complexes. Tusseau-Vuillemin et al. [3, 4], and Ferreira et al. [5], have shown that this fraction is close to the bioavailable fraction for Daphnia magna and aquatic mosses. In addition, DGT is a passive sampling technique, which integrates the metallic contamination during a time deployment of some hours to several weeks. Thus, DGT provides an alternative to spot sampling, which is much more sensitive to concentration fluctuations. For these reasons, passive sampling techniques, and particularly DGT, are gaining acceptance by regulatory and water monitoring authorities.
The main objective of the project presented here was to evaluate the potential of DGT as a monitoring tool on a river basin scale. We present the first results of a large scale deployment of DGT in the Seine river basin. The complete set of measurements constitutes a rich dataset including large and small rivers as well as impacted sites.
Emmanuelle Uher1, Cécile Mirande-Bret2, Catherine Gourlay-Francé1
1. Cemagref , Parc de Tourvoie, 92163 Antony, France
2. LEESU, ENPC, Cité Descartes, 77455 Marne La Vallée, France
Introduction
The link between the chemical speciation of metals and their toxicity [1] has resulted in a number of different analytical techniques to assess the various fractions of metals present in the aqueous environment. Among these, the Diffusive Gradient in Thin-film technique (DGT), developed by Zhang and Davison [2], is commonly used. DGT is an in situ speciation method; it samples a labile fraction composed of free inorganic metals and weakly labile organic complexes. Tusseau-Vuillemin et al. [3, 4], and Ferreira et al. [5], have shown that this fraction is close to the bioavailable fraction for Daphnia magna and aquatic mosses. In addition, DGT is a passive sampling technique, which integrates the metallic contamination during a time deployment of some hours to several weeks. Thus, DGT provides an alternative to spot sampling, which is much more sensitive to concentration fluctuations. For these reasons, passive sampling techniques, and particularly DGT, are gaining acceptance by regulatory and water monitoring authorities.
The main objective of the project presented here was to evaluate the potential of DGT as a monitoring tool on a river basin scale. We present the first results of a large scale deployment of DGT in the Seine river basin. The complete set of measurements constitutes a rich dataset including large and small rivers as well as impacted sites.
|
DGT samples labile metals, which are free or weakly bound to organic matter (Figure 1). Lability is operationally defined i.e. it depends on the thickness and the pore size of the DGT hydrogel. In this study, the use of hydrogels with restricted pore sizes minimized the sampling of large metal complexes.
Conclusion The large scale deployment of DGT in the Seine river basin was successful: results from all the samples have been interpreted. A representative data set of labile metal concentrations in an urban-impacted river basin has been produced. |
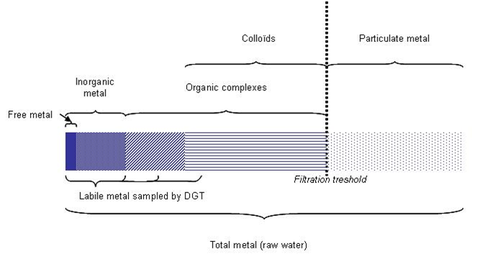
Figure 1: Metal speciation in a water column.
|
References
1. Batley G. E., Apte, S. C.; Stauber, J. L. Speciation and bioavailability of trace metals in water: Progress since 1982. Aust. J. Chem., 2004, 57, 903-919.
2. Zhang, H.; Davison, W. Performance-characteristics of diffusion gradients in thin films for the in situ measurement of trace metals in aqueous solution. Anal. Chem., 1995, 67, 3391-3400.
3. Tusseau-Vuillemin, M.-H.; Gilbin, R.; Bakkaus, E.; Garric, J. Performance of diffusion gradient in thin films to evaluate the toxic fraction of copper to Daphnia magna. Environ. Toxicol. Chem., 2004, 23, 2154-2161.
4. Buzier, R.; Tusseau-Vuillemin, M.-H.; Mouchel, J. M. Evaluation of DGT as a metal speciation tool in wastewater. Sci. Total Environ., 2006, 358, 277-285.
. Ferreira, D.; Tousset, N.; Ridame, C.; Tusseau-Vuillemin, M.-H. More than inorganic copper is bioavailable to aquatic mosses at environmentally relevant concentrations. Environ. Toxicol. Chem., 2008, 27, 2108-2116.
1. Batley G. E., Apte, S. C.; Stauber, J. L. Speciation and bioavailability of trace metals in water: Progress since 1982. Aust. J. Chem., 2004, 57, 903-919.
2. Zhang, H.; Davison, W. Performance-characteristics of diffusion gradients in thin films for the in situ measurement of trace metals in aqueous solution. Anal. Chem., 1995, 67, 3391-3400.
3. Tusseau-Vuillemin, M.-H.; Gilbin, R.; Bakkaus, E.; Garric, J. Performance of diffusion gradient in thin films to evaluate the toxic fraction of copper to Daphnia magna. Environ. Toxicol. Chem., 2004, 23, 2154-2161.
4. Buzier, R.; Tusseau-Vuillemin, M.-H.; Mouchel, J. M. Evaluation of DGT as a metal speciation tool in wastewater. Sci. Total Environ., 2006, 358, 277-285.
. Ferreira, D.; Tousset, N.; Ridame, C.; Tusseau-Vuillemin, M.-H. More than inorganic copper is bioavailable to aquatic mosses at environmentally relevant concentrations. Environ. Toxicol. Chem., 2008, 27, 2108-2116.
Paper 2 On boron in humic and inorganic components of soil: a geochemical vs. a chemical approach
Fyodor S. Kot, Ronit Farran, Malik Kochva, and Avi Shaviv
Department of Environmental, Water and Agricultural Engineering, Technion-Israel Institute of Technology, Haifa 32000, Israel
Introduction
Fyodor S. Kot, Ronit Farran, Malik Kochva, and Avi Shaviv
Department of Environmental, Water and Agricultural Engineering, Technion-Israel Institute of Technology, Haifa 32000, Israel
Introduction
Agulhon (1910) first demonstrated boron is an essential element by observing that boron stimulates the growth of several vascular plants in water culture and that boron is abundant in lignified tissue. Maluga (1964) noticed the boron content of plant ash was much higher than that in soils and the lithosphere (400, 10, and 12 mg kg−1, respectively) indicating that boron accumulates in the biosphere. The accumulation of boron varies in different organisms (Bowen, 1966) (Figure 2)
It seems that boron is not biomagnified in aquatic food chains, but in the terrestrial food chains, boron accumulates in plants but not in animals (Underwood, 1977). A relatively large amount of circumstantial evidence for the essentiality of boron in animals has been appearing since the 1980s (Woods, 1994; Nielsen, 1998).
It seems that boron is not biomagnified in aquatic food chains, but in the terrestrial food chains, boron accumulates in plants but not in animals (Underwood, 1977). A relatively large amount of circumstantial evidence for the essentiality of boron in animals has been appearing since the 1980s (Woods, 1994; Nielsen, 1998).
Reviews on the fate of boron in soil and soil-plant systems reveal some common, but non-attributable, claims. For example, that boron in soil solution is in the form of inorganic boric acid or borates – depending on the environmental pH – and that these forms are the available forms for plant and bacteria utilisation. This assertion probably originates from the inability of many analytical methods to distinguish between organically-bound and inorganic boron complexes.
|
According to Kovda (1973), boron is involved in several major soil processes:
|
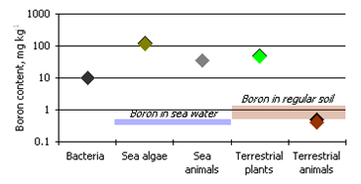
Figure 2 Boron Concentrations in the biosphere
|
Decomposed and humified plants residues are a major source of organic matter in soil, and soil humus is a principal source of nutrients for vegetation. This plant-soil cycle utilises boron.
Some soil chemistry literature claims that soil boron can be subdivided into: boron in soil solution, adsorbed boron, and mineral boron. And the adsorbed boron is considered in the systems of “soil solution-clay minerals” and “iron/aluminium hydro-oxides”. Such an approach seems questionable, and again can be traced to deficiencies in analytical techniques, which neglect boron-organic matter speciation in their procedures (see Kot, 2007, 2009).
Our preliminary results on soil boron fractionation and potential mobility/bioavailability with special reference to soil humus components lead to the following conclusions:
1. Boron in soil is much more organophilic than is often supposed.
2. Humus components are the principal carriers and determinants of boron species in soil.
3. Mobile boron-water species in soils are elated to colloids of a varied, but probably biotic, origin, not simply to inorganic boric acid or borates, as is often presumed.
References for Paper 2
Agulhon, H. (1910). Présence et utilité du bore chez les végétaux. Annales de l’Institut Pasteur (Paris), 24, 321-329.
Bowen, H. J. M. (1966). Trace Elements in Biogeochemistry, Academic Press, London.
Kot, F. S. (2007). Boron speciation in soils irrigated with fresh and effluent municipal waters. Bull. Environ. Contam. Toxicol., 79, 259-263.
Kot, F. S. (2009). Boron sources, speciation and its potential impact on health. Rev. Environ. Sci. Biotechnol., 8, 3-28.
Kovda, V. A. (1973). Fundamentals of Soil Science, Vol. 2, Nauka, Moscow. (In Russian).
Maluga, D. P. (1964). Biogeochemical Methods of Prospecting, Consultants Bureau Plenum Press, New York.
Nielsen, F. H. (1998). Ultratrace elements in nutrition: current knowledge and speculation. J. Trace Elem. Exp. Med., 11, 251-274.
Underwood, E. J. (1977). Trace Elements in Human and Animal Nutrition, Academic Press, New York.
Woods, W.G. (1994). An introduction to boron: history, sources, uses, and chemistry. Environ. Health Perspect., 102, 5-11.
Some soil chemistry literature claims that soil boron can be subdivided into: boron in soil solution, adsorbed boron, and mineral boron. And the adsorbed boron is considered in the systems of “soil solution-clay minerals” and “iron/aluminium hydro-oxides”. Such an approach seems questionable, and again can be traced to deficiencies in analytical techniques, which neglect boron-organic matter speciation in their procedures (see Kot, 2007, 2009).
Our preliminary results on soil boron fractionation and potential mobility/bioavailability with special reference to soil humus components lead to the following conclusions:
1. Boron in soil is much more organophilic than is often supposed.
2. Humus components are the principal carriers and determinants of boron species in soil.
3. Mobile boron-water species in soils are elated to colloids of a varied, but probably biotic, origin, not simply to inorganic boric acid or borates, as is often presumed.
References for Paper 2
Agulhon, H. (1910). Présence et utilité du bore chez les végétaux. Annales de l’Institut Pasteur (Paris), 24, 321-329.
Bowen, H. J. M. (1966). Trace Elements in Biogeochemistry, Academic Press, London.
Kot, F. S. (2007). Boron speciation in soils irrigated with fresh and effluent municipal waters. Bull. Environ. Contam. Toxicol., 79, 259-263.
Kot, F. S. (2009). Boron sources, speciation and its potential impact on health. Rev. Environ. Sci. Biotechnol., 8, 3-28.
Kovda, V. A. (1973). Fundamentals of Soil Science, Vol. 2, Nauka, Moscow. (In Russian).
Maluga, D. P. (1964). Biogeochemical Methods of Prospecting, Consultants Bureau Plenum Press, New York.
Nielsen, F. H. (1998). Ultratrace elements in nutrition: current knowledge and speculation. J. Trace Elem. Exp. Med., 11, 251-274.
Underwood, E. J. (1977). Trace Elements in Human and Animal Nutrition, Academic Press, New York.
Woods, W.G. (1994). An introduction to boron: history, sources, uses, and chemistry. Environ. Health Perspect., 102, 5-11.
Paper 3 Impact of metals on the biodegradation of polycyclic aromatic hydrocarbons in soil
Ifeyinwa S. Obuekwe and Kirk T. Semple
Lancaster Environmental Centre, Lancaster University, UK
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are organic substances which contain two or more fused aromatic rings and are common in a wide range of petroleum products, as well as crude oil (Bamforth and Singleton, 2005; Wong et al., 2005). They are by-products from incomplete combustion of organic substances such as coal, oil, petrol, and wood, and are ubiquitous in the soil environment (Sokhn et al., 2001; Maliszewska-Kordybach and Smreczak, 2003). While concentrations of individual PAHs in soil produced by natural processes are estimated to be around 1-10 µg/kg, their concentrations in highly polluted soils vary from 10 mg/kg to 10 g/kg dry weight (Sokhn et al., 2001). PAHs are highly recalcitrant molecules that can persist in soil due to their hydrophobicity and low water solubility, and because some are known to be mutagenic, teratogenic and carcinogenic their fate in nature is therefore of environmental concern. (Sokhn et al., 2001; Bamforth and Singleton, 2005).
The persistence of PAHs in soil depends on a variety of factors, for example the chemical structure of the PAH, the concentration and dispersion of the PAH, and the bioavailability of the contaminant. Soil type and structure, pH, temperature, nutrients and water for the activity of the pollutant-degrading microbial community, will all influence the time that PAHs persist in soil (Bamforth and Singleton, 2005).
The association of PAHs with co-pollutants such as heavy metals will also prolong PAH residence time in soil. The presence of heavy metals in soil could inhibit microbial growth and hence limit the metabolism of PAHs in soil.
PAHs can be biologically degraded and removed from the natural environment and/or converted into less harmful products by the indigenous microbial community present in a contaminated environment (Bamforth and Singleton, 2005). PAH-degrading microorganisms are ubiquitously distributed in the natural environment, such as soils. These include bacteria such as the genus Pseudomonas and Rhodococcus as well as fungi such as Chrysosporium and Aspergillus. However, the presence of heavy metals in soil could adversely affect microflora populations and hence PAH biodegradation.
Metals are ubiquitous in nature and even those metals generally considered as pollutants are found in trace concentrations in the soil environment. Such metals include mercury, lead, arsenic, cadmium, chromium, manganese which are commonly associated with pollution and toxicity, but also include elements (e.g. Zn, Cu and Ni) which are essential in the metabolism of living organisms, albeit at low concentrations (Wong et al., 2005). Mining and ore refinement, nuclear processing, and industrial manufacture of a variety of products such as batteries, metal alloys, and fungicides are some of the sources of these metals in the soil environment. These metals in soil can exist as individual metals or more often as metal mixtures and their elevated concentrations in the soil environment have wide-ranging impacts on animal, plant, and microbial species (microbial growth is often slowed or inhibited completely in the presence of excessive amounts of metals) (Roane et al., 2005). Due to their toxic nature, the presence of metals in organic-contaminated sites often complicates and limits the bioremediation process.
PAHs and heavy metals are among mixed pollution of major concerns in the soil environment because of the risk they pose to soil health. Soils contaminated with PAHs often contain other pollutants such as heavy metals (HM) this is because often, these contaminants come from same sources (Maliszewska-Kordybach and Smreczak, 2003). Sources of these pollutants in soil include atmospheric deposition, power and heat generation, old gasification and wood preserving plants, metallurgical industries, and fungicide application.
Fate and behaviour of PAHs and metals in soil
Diverse pathways in the soil environment influence the fate and behaviour of both PAHs and metals. PAHs in soil can volatilize, be leached, complex with soil minerals and organic matter, biodegrade to carbon dioxide, or undergo carbon assimilation by the microbial biomass (Figure 3). Metals can be removed by volatilization, and when in an aqueous phase, losses can be a result of plant uptake, surface runoff, complexation or sorption to minerals and organic matter, and precipitation of the metal salt
Ifeyinwa S. Obuekwe and Kirk T. Semple
Lancaster Environmental Centre, Lancaster University, UK
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are organic substances which contain two or more fused aromatic rings and are common in a wide range of petroleum products, as well as crude oil (Bamforth and Singleton, 2005; Wong et al., 2005). They are by-products from incomplete combustion of organic substances such as coal, oil, petrol, and wood, and are ubiquitous in the soil environment (Sokhn et al., 2001; Maliszewska-Kordybach and Smreczak, 2003). While concentrations of individual PAHs in soil produced by natural processes are estimated to be around 1-10 µg/kg, their concentrations in highly polluted soils vary from 10 mg/kg to 10 g/kg dry weight (Sokhn et al., 2001). PAHs are highly recalcitrant molecules that can persist in soil due to their hydrophobicity and low water solubility, and because some are known to be mutagenic, teratogenic and carcinogenic their fate in nature is therefore of environmental concern. (Sokhn et al., 2001; Bamforth and Singleton, 2005).
The persistence of PAHs in soil depends on a variety of factors, for example the chemical structure of the PAH, the concentration and dispersion of the PAH, and the bioavailability of the contaminant. Soil type and structure, pH, temperature, nutrients and water for the activity of the pollutant-degrading microbial community, will all influence the time that PAHs persist in soil (Bamforth and Singleton, 2005).
The association of PAHs with co-pollutants such as heavy metals will also prolong PAH residence time in soil. The presence of heavy metals in soil could inhibit microbial growth and hence limit the metabolism of PAHs in soil.
PAHs can be biologically degraded and removed from the natural environment and/or converted into less harmful products by the indigenous microbial community present in a contaminated environment (Bamforth and Singleton, 2005). PAH-degrading microorganisms are ubiquitously distributed in the natural environment, such as soils. These include bacteria such as the genus Pseudomonas and Rhodococcus as well as fungi such as Chrysosporium and Aspergillus. However, the presence of heavy metals in soil could adversely affect microflora populations and hence PAH biodegradation.
Metals are ubiquitous in nature and even those metals generally considered as pollutants are found in trace concentrations in the soil environment. Such metals include mercury, lead, arsenic, cadmium, chromium, manganese which are commonly associated with pollution and toxicity, but also include elements (e.g. Zn, Cu and Ni) which are essential in the metabolism of living organisms, albeit at low concentrations (Wong et al., 2005). Mining and ore refinement, nuclear processing, and industrial manufacture of a variety of products such as batteries, metal alloys, and fungicides are some of the sources of these metals in the soil environment. These metals in soil can exist as individual metals or more often as metal mixtures and their elevated concentrations in the soil environment have wide-ranging impacts on animal, plant, and microbial species (microbial growth is often slowed or inhibited completely in the presence of excessive amounts of metals) (Roane et al., 2005). Due to their toxic nature, the presence of metals in organic-contaminated sites often complicates and limits the bioremediation process.
PAHs and heavy metals are among mixed pollution of major concerns in the soil environment because of the risk they pose to soil health. Soils contaminated with PAHs often contain other pollutants such as heavy metals (HM) this is because often, these contaminants come from same sources (Maliszewska-Kordybach and Smreczak, 2003). Sources of these pollutants in soil include atmospheric deposition, power and heat generation, old gasification and wood preserving plants, metallurgical industries, and fungicide application.
Fate and behaviour of PAHs and metals in soil
Diverse pathways in the soil environment influence the fate and behaviour of both PAHs and metals. PAHs in soil can volatilize, be leached, complex with soil minerals and organic matter, biodegrade to carbon dioxide, or undergo carbon assimilation by the microbial biomass (Figure 3). Metals can be removed by volatilization, and when in an aqueous phase, losses can be a result of plant uptake, surface runoff, complexation or sorption to minerals and organic matter, and precipitation of the metal salt
|
Ageing of PAHs and metals in soil is a major factor in determining their bioavailability, because of the affinity these co-contaminants have for soil organic matter and clay content. In well-aged soils, mild extractability, bioavailability and biodegradation assays for PAHs are usually low (Stokes et al., 2005). Ageing of metals in soils has been demonstrated to be a major factor in determining their bioavailability or lability, with bioavailability decreasing with increases in metal-soil contact times (Wendling, 2009). Several processes contribute to metal ageing in soils including incorporation into mineral structures, diffusion into pore spaces within minerals, nucleation or precipitation, mineral surface oxidation and entrainment via the formation of chemical complexes with soil solids (Wendling et al., 2009).
|
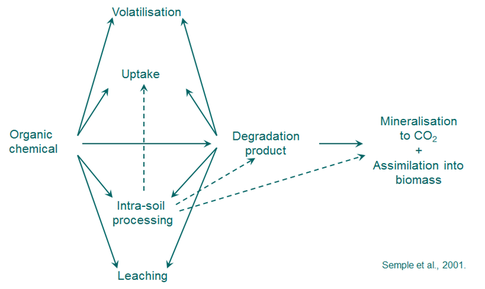
Figure 3 Fate and behaviour of an organic chemical (e.g. a PAH) in soil
|
Metal toxicity to the microbial cell
Metals are essential components of microbial cells; for example, sodium (Na) and potassium (K) regulate gradients across the cell membrane, and copper, iron and manganese are required for the activity of key metalloenzymes in photosynthesis and electron transport (Roane et al., 2005). However, metals can also be extremely toxic to microorganisms (especially at high concentrations) impacting on microbial growth, morphology and biochemical activities as a result of specific interactions with cellular components (Giller et al., 1998; Wong et al., 2005). This toxicity of metals towards microorganisms affects the degradation of PAHs in soil.
Metals are essential components of microbial cells; for example, sodium (Na) and potassium (K) regulate gradients across the cell membrane, and copper, iron and manganese are required for the activity of key metalloenzymes in photosynthesis and electron transport (Roane et al., 2005). However, metals can also be extremely toxic to microorganisms (especially at high concentrations) impacting on microbial growth, morphology and biochemical activities as a result of specific interactions with cellular components (Giller et al., 1998; Wong et al., 2005). This toxicity of metals towards microorganisms affects the degradation of PAHs in soil.
|
Mechanisms of metal toxicity differ. Toxicity may occur as a result of binding of the metal to ligands containing sulfhydryl, carboxyl or phosphate groups such as proteins or nucleic acids (Roane et al., 2005). Mercury (Hg) and cadmium (Cd) cations readily bind sulfhydryl groups causing protein synthesis inhibition (Figure 4). It could also be as a result of metal-catalyzed decomposition of essential metabolites and analogue replacement of structurally important cell components. A good example is arsenic (As), which is bactericidal because it acts as an analogue of phosphate, disrupting nucleic acid structure and enzyme action (Figure 4) (Roane et al., 2005). High concentrations of lead (Pb) and nickel (Ni) retard cell division, while copper (Cu) and zinc (Zn) cause cell membrane disruption (Figure 4).
|
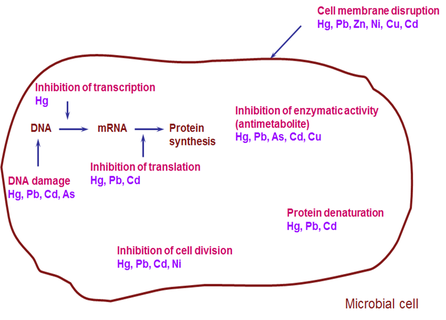
Figure 4 Mechanisms of metal toxicity to a microbial cell.
|
A wide range of soil properties such as pH, organic matter content, clay content, iron oxide content and redox potential affect soil metal concentrations and hence the impact of metals on soil microbes (Giller et al., 1998). Of these, pH has the greatest influence because pH determines the solubility and speciation of metals in soil. The free metal ion is generally assumed to be the chemical species which is taken up by soil microbes and is toxic when present in an excess (Giller et al., 1998).
These negative effects of heavy metals on soil microbes and soil microbial processes can potentially limit the bioremediation of organic pollutants. Examples from the literature include:
Methodologies for studying PAHs and metals in the laboratory
Techniques for studying soils contaminated with PAHs and metals in the laboratory examine:
(a) PAH degradation (b) microbial activity (respiration, enzyme activity etc.) and (c) microbial population. The PAH–metal(s) co-contaminant mixtures may be either artificially simulated in the laboratory or soils already contaminated with the co-contaminants in the environment are used. The co-contaminants are incubated in the presence of indigenous microflora or introduced inocula for a period of time after which PAH degradation, microbial activity as well as bacteria count and/or fungal soil colonization are assessed.
These negative effects of heavy metals on soil microbes and soil microbial processes can potentially limit the bioremediation of organic pollutants. Examples from the literature include:
- Decreases in phenanthrene degradation, microbial respiration and microbial numbers with increasing concentrations of Cu (Sokhn et al., 2001; Obuekwe and Semple, 2011).
- Inhibition of phenanthrene biodegradation and microbial metabolic activity at high Zn concentrations (Wong et al., 2005).
- Decrease in PAH biodegradation and mycelial soil colonization in Cd–PAH–contaminated soils (Baldrin et al., 2000).
- Lower microbial count and carbon dioxide evolution in soil in the presence of PAHs and cupric sulphate (CuSO4). This is thought to be as a result of inhibition of microbial growth and metabolism in the presence of CuSO4 (Atagana, 2006).
Methodologies for studying PAHs and metals in the laboratory
Techniques for studying soils contaminated with PAHs and metals in the laboratory examine:
(a) PAH degradation (b) microbial activity (respiration, enzyme activity etc.) and (c) microbial population. The PAH–metal(s) co-contaminant mixtures may be either artificially simulated in the laboratory or soils already contaminated with the co-contaminants in the environment are used. The co-contaminants are incubated in the presence of indigenous microflora or introduced inocula for a period of time after which PAH degradation, microbial activity as well as bacteria count and/or fungal soil colonization are assessed.
Techniques for analysing PAH degradation include: solvent extraction and GC (Sokhn et al., 2001); solvent extraction and GC-MS (Baldrin et al., 2000; Wong et al., 2005); conversion of 14C PAH into 14CO2 using respirometry (Semple et al., 2001; Obuekwe and Semple, 2011). Microbial activity can be assessed by measuring CO2 evolution with an infrared CO2 analyser (Sokhn et al., 2001) or 14CO2 evolution using respirometry (Semple et al., 2001; Obuekwe and Semple, 2011). Microbial metabolic activity can also be determined by measuring various enzyme activities (Wong et al., 2005; Baldrin et al., 2000). Microbial populations may be examined using a bacteria plate count technique (Sokhn et al., 2001; Atagana 2006; Obuekwe and Semple, 2011). Fungal soil colonization can be used to assess fungal soil growth (Baldrin et al., 2000).
Factors that influence the impact of metals on PAH biodegradation in soil
Soil type: Soil pH has the highest influence on metal toxicity because of its impact on metal solubility and speciation in soil. Soils with low pH usually contain metals in a more soluble form resulting in enhanced metal bioavailability and toxicity. For example, Wendling et al., (2009) found an increase in isotopically exchangeable cobalt in acidic soils compared with neutral or alkaline pH. Biodegradation of PAHs in the presence of metals in soil with a low pH could therefore be impeded.
Organic matter content, clay content, and cation exchange capacity (CEC) are other soil properties that affect metal toxicity. Low lability of Cu in soils with high organic matter has been reported (Ma et al., 2006). Finer textured soil, which is much higher in clay content and cation exchange capacity, possesses a greater binding ability for added metals than coarse-textured soil (Kim et al., 2008).
Metal ageing in soil: Ageing of metals in soils is a major factor in determining their bioavailability and toxicity, with metal bioavailability decreasing as metal-soil contact time increases. For example, a decrease in the readily soluble and weakly sorbed Co pools in soils with time has been reported (Wendling et al., 2009). Recent unpublished research (Obuekwe and Semple, 2011) on the impact of Zn or Cu on phenanthrene catabolism in soil showed significant increases in phenanthrene catabolism at high Cu concentration (500 mg/kg) with time [also vide infra]. This could be as a result of Cu diffusing into nanopores or interstices and becoming less bioavailable to prevent phenanthrene catabolism in soil.
Metal concentration in soil: Heavy metals are known to be potentially toxic to soil microorganisms at high concentrations and can hinder the biodegradation of organic contaminant in soil (Wong et al., 2005). Many studies have reported an increase in metal toxicity with increasing concentration; Cu at concentrations of 700 and 7000 mg/kg soil were found to reduce microbial activity and phenanthrene biodegradation (Sokhn et al., 2001). Raised Zn concentrations in soil (720 and 1440 mg/kg) inhibited both phenanthrene biodegradation rates and microbial metabolic activity. High Cu concentrations (500 and 1000 mg/kg) reduced microbial numbers and phenanthrene biodegradation in soil (Obuekwe and Semple, 2011) [also vide supra].
Soil type: Soil pH has the highest influence on metal toxicity because of its impact on metal solubility and speciation in soil. Soils with low pH usually contain metals in a more soluble form resulting in enhanced metal bioavailability and toxicity. For example, Wendling et al., (2009) found an increase in isotopically exchangeable cobalt in acidic soils compared with neutral or alkaline pH. Biodegradation of PAHs in the presence of metals in soil with a low pH could therefore be impeded.
Organic matter content, clay content, and cation exchange capacity (CEC) are other soil properties that affect metal toxicity. Low lability of Cu in soils with high organic matter has been reported (Ma et al., 2006). Finer textured soil, which is much higher in clay content and cation exchange capacity, possesses a greater binding ability for added metals than coarse-textured soil (Kim et al., 2008).
Metal ageing in soil: Ageing of metals in soils is a major factor in determining their bioavailability and toxicity, with metal bioavailability decreasing as metal-soil contact time increases. For example, a decrease in the readily soluble and weakly sorbed Co pools in soils with time has been reported (Wendling et al., 2009). Recent unpublished research (Obuekwe and Semple, 2011) on the impact of Zn or Cu on phenanthrene catabolism in soil showed significant increases in phenanthrene catabolism at high Cu concentration (500 mg/kg) with time [also vide infra]. This could be as a result of Cu diffusing into nanopores or interstices and becoming less bioavailable to prevent phenanthrene catabolism in soil.
Metal concentration in soil: Heavy metals are known to be potentially toxic to soil microorganisms at high concentrations and can hinder the biodegradation of organic contaminant in soil (Wong et al., 2005). Many studies have reported an increase in metal toxicity with increasing concentration; Cu at concentrations of 700 and 7000 mg/kg soil were found to reduce microbial activity and phenanthrene biodegradation (Sokhn et al., 2001). Raised Zn concentrations in soil (720 and 1440 mg/kg) inhibited both phenanthrene biodegradation rates and microbial metabolic activity. High Cu concentrations (500 and 1000 mg/kg) reduced microbial numbers and phenanthrene biodegradation in soil (Obuekwe and Semple, 2011) [also vide supra].
Metal speciation: The potential toxicity of a heavy metal in soil depends upon its speciation and availability. Heavy metals are present in various forms with different degrees of mobility and bioavailability. The toxicity of a heavy metal can be linked to its free-ion activity (Chaperon and Sauve, 2008; Kim et al., 2008). Kim et al., (2008) provided evidence that Cu2+ was a better surrogate for estimating the toxic effect of Cu than Cu extractable by CaCl2. Free Cu2+ ion is the likely toxic species of Cu, reacting directly with sulfhydryl groups on the active site of the enzyme (dehydrogenase) and causing its inhibition. Metal speciation in soil is influenced by a number of physico-chemical properties including soil pH, clay and organic matter content, Fe and Mn oxide composition and content, as well as long term fixation or ageing effects (Wendling et al., 2009).
Conclusion
Heavy metals at elevated concentrations in the soil environment have a range of effects on microbial species. These include a decrease in microbial count and disruption of nucleic acid and enzyme function, which subsequently reduce or impede the biodegradation of PAHs in soil. Heavy metal toxicity in soil depends on factors which affect their interaction with PAHs. Due to their toxic nature, the presence of metals in PAH–contaminated land sites can often complicate and limit bioremediation processes.
References for Paper 3
Atagana, H. I. (2006). Biodegradation of polycyclic aromatic hydrocarbons in contaminated soil by biostimulation and bioaugmentation in the presence of copper (II) ions. World J. Microbiol. Biotechnol., 22, 1145-1153.
Baldrin, P.; Wiesche, C.; Gabriel, J.; Nerud, F.; Zadrazil, F. (2000). Influence of cadmium and mercury on activities of ligninolytic enzymes and degradation of polycyclic aromatic hydrocarbons by Pleurotus osreatus in soil. Appl. Environ. Microbiol., 66, 2471-2478.
Bamforth, S. M.; Singleton L. (2005). Bioremediation of polycyclic aromatic hydrocarbons: current knowledge and future directions. J. Chem. Technol. Biotechnol., 80, 723-736.
Chaperon, S.; Sauve, S. (2008). Toxicity interactions of cadmium, copper, and lead on soil urease and dehydrogenase activity in relation to chemical speciation. Ecotoxicol. Environ. Saf., 70, 1-9.
Giller, K. E.; Witter, E.; Mcgrath, S. P. (1998). Toxicity of heavy metals to microorganisms and microbial processes in agricultural soils: a review. Soil Biol. Biochem., 30, 1389-1414.
Kim, B.; Mcbride, M. B.; Hay, A. G. (2008). Urease activity in aged copper and zinc-spiked soils: relationship to CaCl2–extractable metals and Cu2+ activity. Environ. Toxicol. Chem., 27, 2469-2475.
Ma, Y.; Lombi, E.; Nolan, A. L.; Mclaughlin, M. J. (2006). Short-term natural attenuation of copper in soils: effects of time, temperature and soil characteristics. Environ. Toxicol. Chem., 25, 652-658.
Maliszewska-Kordybach, B.; Smreczak, B., (2003). Habitat function of agricultural soils as affected by heavy metals and polycyclic aromatic hydrocarbons contamination. Environ. Int., 28, 719-728.
Obuekwe, I. S.; Semple, K. T., Impact of Zn or Cu-phenanthrene contamination on phenanthrene catabolism in soil. (2011). Manuscript in preparation.
Roane, T. M.; Pepper, I. L.; Miller, R. M. (2005). Microbial remediation of metals. In Bioremediation Principles and Applications, Crawford R.L.; Crawford D.L. (eds.), Cambridge University Press, Cambridge, pp 312-340.
Semple, K.T.; Reid, B. J.; Fermor, T. R. (2001). Impact of composting strategies on the treatment of soils contaminated with organic pollutants. Environ. Pollut., 112, 269-283.
Sokhn, J.; De Leij, F.A. A. M.; Hart, T. D.; Lynch, J. M. (2001). Effect of copper on the degradation of phenanthrene by soil microorganisms. Lett. Appl. Microbiol., 33, 164-168.
Stokes, J. D.; Paton, G. I.; Semple, K. T. (2005). Behaviour and assessment of bioavailability of organic contaminants in soil: relevance for risk assessment and remediation, soil use and management. 21, 475-486
Wendling, L. A.; Kirby, J. K.; McLaughlin, M. J. (2009). Aging effects on cobalt availability in soils. Environ. Toxicol. Chem., 28, 1609-17.
Wong, K. W.; Toh, B. A.; Ting, Y. P.; Obbard, J. P. (2005). Biodegradation of phenanthrene by the indigenous microbial biomass in a zinc amended soil. Lett. Appl. Microbiol., 40, 50-55.
Heavy metals at elevated concentrations in the soil environment have a range of effects on microbial species. These include a decrease in microbial count and disruption of nucleic acid and enzyme function, which subsequently reduce or impede the biodegradation of PAHs in soil. Heavy metal toxicity in soil depends on factors which affect their interaction with PAHs. Due to their toxic nature, the presence of metals in PAH–contaminated land sites can often complicate and limit bioremediation processes.
References for Paper 3
Atagana, H. I. (2006). Biodegradation of polycyclic aromatic hydrocarbons in contaminated soil by biostimulation and bioaugmentation in the presence of copper (II) ions. World J. Microbiol. Biotechnol., 22, 1145-1153.
Baldrin, P.; Wiesche, C.; Gabriel, J.; Nerud, F.; Zadrazil, F. (2000). Influence of cadmium and mercury on activities of ligninolytic enzymes and degradation of polycyclic aromatic hydrocarbons by Pleurotus osreatus in soil. Appl. Environ. Microbiol., 66, 2471-2478.
Bamforth, S. M.; Singleton L. (2005). Bioremediation of polycyclic aromatic hydrocarbons: current knowledge and future directions. J. Chem. Technol. Biotechnol., 80, 723-736.
Chaperon, S.; Sauve, S. (2008). Toxicity interactions of cadmium, copper, and lead on soil urease and dehydrogenase activity in relation to chemical speciation. Ecotoxicol. Environ. Saf., 70, 1-9.
Giller, K. E.; Witter, E.; Mcgrath, S. P. (1998). Toxicity of heavy metals to microorganisms and microbial processes in agricultural soils: a review. Soil Biol. Biochem., 30, 1389-1414.
Kim, B.; Mcbride, M. B.; Hay, A. G. (2008). Urease activity in aged copper and zinc-spiked soils: relationship to CaCl2–extractable metals and Cu2+ activity. Environ. Toxicol. Chem., 27, 2469-2475.
Ma, Y.; Lombi, E.; Nolan, A. L.; Mclaughlin, M. J. (2006). Short-term natural attenuation of copper in soils: effects of time, temperature and soil characteristics. Environ. Toxicol. Chem., 25, 652-658.
Maliszewska-Kordybach, B.; Smreczak, B., (2003). Habitat function of agricultural soils as affected by heavy metals and polycyclic aromatic hydrocarbons contamination. Environ. Int., 28, 719-728.
Obuekwe, I. S.; Semple, K. T., Impact of Zn or Cu-phenanthrene contamination on phenanthrene catabolism in soil. (2011). Manuscript in preparation.
Roane, T. M.; Pepper, I. L.; Miller, R. M. (2005). Microbial remediation of metals. In Bioremediation Principles and Applications, Crawford R.L.; Crawford D.L. (eds.), Cambridge University Press, Cambridge, pp 312-340.
Semple, K.T.; Reid, B. J.; Fermor, T. R. (2001). Impact of composting strategies on the treatment of soils contaminated with organic pollutants. Environ. Pollut., 112, 269-283.
Sokhn, J.; De Leij, F.A. A. M.; Hart, T. D.; Lynch, J. M. (2001). Effect of copper on the degradation of phenanthrene by soil microorganisms. Lett. Appl. Microbiol., 33, 164-168.
Stokes, J. D.; Paton, G. I.; Semple, K. T. (2005). Behaviour and assessment of bioavailability of organic contaminants in soil: relevance for risk assessment and remediation, soil use and management. 21, 475-486
Wendling, L. A.; Kirby, J. K.; McLaughlin, M. J. (2009). Aging effects on cobalt availability in soils. Environ. Toxicol. Chem., 28, 1609-17.
Wong, K. W.; Toh, B. A.; Ting, Y. P.; Obbard, J. P. (2005). Biodegradation of phenanthrene by the indigenous microbial biomass in a zinc amended soil. Lett. Appl. Microbiol., 40, 50-55.
Paper 4 Characterisation of soils containing naturally occurring radioactive material (NORM) using in situ and rapid laboratory techniques. Case study: South Terras, Cornwall.
Anna Kutner (1), Dr Stuart Black (1), Dr Helen Beddow (2), Dr Matthew Almond (1)
1. University of Reading; 2. Nuvia, Harwell, Oxford
Introduction
In situ analysis has become increasingly important for contaminated land investigation and remediation. At present, portable techniques are used mainly as scanning tools to assess the spread and magnitude of the contamination, and are an adjunct to conventional laboratory analyses. A site in Cornwall, containing naturally occurring radioactive material (NORM), provided an opportunity for Reading University PhD student Anna Kutner to compare analytical data collected in situ with data generated by laboratory-based methods. The preliminary results in this paper extend the author’s poster presentation at last September’s GeoSpec2010 conference held in Lancaster.
Case study site: South Terras, Cornwall
South Terras is a historical mining site located to the west from St Austell in the valley of River Fal, Cornwall. The area has a strictly metaliferrous character, located directly on a tin and uranium vein, hence there is a long history of mining activities, which can be roughly divided into: iron–tin mining (1870–1890s), uranium mining (1890s–1910s), and radium extraction works (1910s–1930s) [1, 2]. After closure in the early 1930s, the site has been left undisturbed and currently has a SSSI status (Sites of Special Scientific Interest) due to the occurrence of rare minerals. Consequently South Terras provides an ideal environment for research on the behaviour of natural radionuclides in a post-industrial environment. [See also http://www.dangerouslaboratories.org/rcw3.html for further details of the location].
Anna Kutner (1), Dr Stuart Black (1), Dr Helen Beddow (2), Dr Matthew Almond (1)
1. University of Reading; 2. Nuvia, Harwell, Oxford
Introduction
In situ analysis has become increasingly important for contaminated land investigation and remediation. At present, portable techniques are used mainly as scanning tools to assess the spread and magnitude of the contamination, and are an adjunct to conventional laboratory analyses. A site in Cornwall, containing naturally occurring radioactive material (NORM), provided an opportunity for Reading University PhD student Anna Kutner to compare analytical data collected in situ with data generated by laboratory-based methods. The preliminary results in this paper extend the author’s poster presentation at last September’s GeoSpec2010 conference held in Lancaster.
Case study site: South Terras, Cornwall
South Terras is a historical mining site located to the west from St Austell in the valley of River Fal, Cornwall. The area has a strictly metaliferrous character, located directly on a tin and uranium vein, hence there is a long history of mining activities, which can be roughly divided into: iron–tin mining (1870–1890s), uranium mining (1890s–1910s), and radium extraction works (1910s–1930s) [1, 2]. After closure in the early 1930s, the site has been left undisturbed and currently has a SSSI status (Sites of Special Scientific Interest) due to the occurrence of rare minerals. Consequently South Terras provides an ideal environment for research on the behaviour of natural radionuclides in a post-industrial environment. [See also http://www.dangerouslaboratories.org/rcw3.html for further details of the location].
|
Methodology
Techniques selected to assess the South Terras site are shown in Table 1. The techniques aim to determine: the distribution of gamma-emitting radionuclides at the site (Groundhog), characterise the nature of NORM contamination present at the site/in the samples (gamma spectroscopy), radon in ambient air (RAD7), composition of the surface of buildings still remaining at the site (PXRF, excluding radium), elemental composition (XRF, ICP-OES) and mineralogy of the topsoil (XRD, IR) based on the samples collected during the first field trip in April 2010. An additional aspect of this project will involve a comparison of the methods employed in terms of their accuracy and precision, duration of analysis, and portability. |

Table 1 The range of techniques employed to assess the South Terras mining site
|
Preliminary observations and results
Radon measurements were conducted throughout the site, which established that the average concentration of radon in the ambient air was between 100-300 Bq m-3. However, in the northern corner of the site the radon exhalation reached a level of 16,000 Bq m-3 (using RAD7). This is likely to be an indication of the location of the Northern Shaft, not recorded on current OS maps (Figure 5, Mark A). The Geiger counter proved to be a useful tool to roughly indentify “hot spots”, especially in the woodlands where the GPS signal used by the Groundhog system was lost. The areas of elevated radiation correlated perfectly with the historical location of the ore processing and extracting units (data for those finding will be reported at a later date). The buildings located at the site still contain some level of surface contamination. The highest readings between 550-800 cps were recorded for the walls of Building A (Figure 5, Mark B) (background at the site ~40-60 cps). The building was considered to be an office/storage unit, however the concentration of U above 200 ppm (PXRF) indicated that the building material used for construction contained materials enriched in uranium and thorium, possibly the spoil material from the industrial processes at the site. Slime tanks (Figure 5, Mark C) located behind Building A contain radium-contaminated waste deposited there after the closure in 1930s. It is assumed that waste slurry was mixed with wet concrete and covered with a concrete slab for better shielding. Uranium levels on the surface of the slab were below LOD (PXRF), but a radon peak was recorded (>270 Bq m-3), which might indicate imperfections in the concrete structure (e.g. formation of cracks) or improper mixing application and emanation of radon from the decay of radium contained in waste.
Radon measurements were conducted throughout the site, which established that the average concentration of radon in the ambient air was between 100-300 Bq m-3. However, in the northern corner of the site the radon exhalation reached a level of 16,000 Bq m-3 (using RAD7). This is likely to be an indication of the location of the Northern Shaft, not recorded on current OS maps (Figure 5, Mark A). The Geiger counter proved to be a useful tool to roughly indentify “hot spots”, especially in the woodlands where the GPS signal used by the Groundhog system was lost. The areas of elevated radiation correlated perfectly with the historical location of the ore processing and extracting units (data for those finding will be reported at a later date). The buildings located at the site still contain some level of surface contamination. The highest readings between 550-800 cps were recorded for the walls of Building A (Figure 5, Mark B) (background at the site ~40-60 cps). The building was considered to be an office/storage unit, however the concentration of U above 200 ppm (PXRF) indicated that the building material used for construction contained materials enriched in uranium and thorium, possibly the spoil material from the industrial processes at the site. Slime tanks (Figure 5, Mark C) located behind Building A contain radium-contaminated waste deposited there after the closure in 1930s. It is assumed that waste slurry was mixed with wet concrete and covered with a concrete slab for better shielding. Uranium levels on the surface of the slab were below LOD (PXRF), but a radon peak was recorded (>270 Bq m-3), which might indicate imperfections in the concrete structure (e.g. formation of cracks) or improper mixing application and emanation of radon from the decay of radium contained in waste.
|
Mineralogy of the site comprises quartz (SiO2) and hematite (Fe2O3), as well as calcite (CaCO3) and aluminosilicates, especially kaolinite, the presence of which is to be expected considering the proximity of china clay located just a few miles north from South Terras, where mineral used to be excavated for production of porcelain and, more recently, fine grade paper. Samples have high organic content which might provide a natural sink for some radionuclides due to complexing properties of humic and fulvic acids [4, 5].
Discussion Due to the local geology, the study area is naturally enriched in U and Th. However, industrial activity has exposed the surface soils to high-grade uranium ore (approx. 36%) [1] comprising pitchblende and uraninite (UO2-U3O8), as well as the products of ore extraction and processing procedures. As expected, the samples collected in the vicinity of extraction and smelting facilities were enriched in radionuclides from 238U and 232Th decay chains, all levels within the limits set by Schedule 23 in the Environmental Permitting regulations (2010) (previously Schedule 1 in the Radioactive Substances Act 1993). |
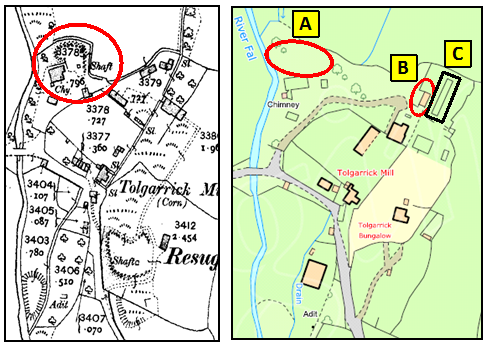
Figure 5 Detailed maps of South Terras from 1907 (left) and 2010 (right marked with "hot spots") [3]
|
The presence of calcite (CaCO3) and hematite (Fe2O3) at the site might indicate a potential for entrapment of radium bivalent ion Ra2+ by substitution for calcium in calcite due to common origin as earth alkaline elements or formation of insoluble complexes with iron oxides [6]. Uranyl ion (UO22+) forms highly soluble complexes with carbonates over a wide range of pH, thus there is a potential for leaching of U into groundwater or transport in the run-off to the nearby River Fal.
However, on the entry points to the River Fal no increase in the concentration of uranium in water has been observed, since the main fraction of the radionuclide is bound to the sediments [7]. Additionally, the enhanced presence of organic matter is another potential mechanism for immobilisation of radionuclides due to the complexing properties of humic and fulvic acids. Those properties are being investigated more fully as a part of the research about the construction of a deep geological repository in United Kingdom to store intermediate and high level waste [4, 5].
Summary
This paper presents our preliminary findings at South Terras. During the early part of 2011 we will complete the data acquisition and interpretation to provide a fuller picture of this site. These results will be complemented by historical data obtained from the Cornwall Record Office on the type of extraction techniques employed at South Terras from ca. 1870–1930.
Acknowledgments
I thank Nuvia for the loan of the GroundhogTM system and Dr Black and Dr Almond for their help during the first field trip. I am grateful to the Boconnoc Estate for allowing me access to South Terras, and I thank Dr Imad Ahmed for inviting me to submit this article to the ECG Bulletin.
References for Paper 4
1. Smale, C. V. South Terras: Cornwall’s premier uranium and radium mine. Journal of the Royal Institution of Cornwall. New Series, 1993, 1(3), 304-321.
2. Dines, H. G. The metalliferous mining region of SW England. Volume 2. Memoirs of the Geological Survey of Great Britain (England and Wales), 1956, HMSO, London.
3. EDINA, South Terras. 2010, Digimap Collection.
4. Keith-Roach, M. J. The speciation, stability, solubility and biodegradation of organic co-contaminant radionuclide complexes: a review. Sci. Total Environ., 2008, 396, 1-1
5. Carlsen, L. The Role of Organics on the Migration of Radionuclides in the Geosphere: Final Report, Nuclear Science and Technology, Commission of the European Communities, Luxembourg, 1989.
6. Baker, A. C.; Toque, C. A review of the potential for radium from luminising activities to migrate in the environment. J. Radiol. Prot., 2005, 25, 127-140.
7. Moliner-Martinez, Y. Campíns-Falcó. P.; Worsfold, P. J.; Keith-Roach, M. J. The impact of a disused mine on uranium transport in the River Fal, South West England. J. Environ. Monit., 2004, 6, 907-913.
Summary
This paper presents our preliminary findings at South Terras. During the early part of 2011 we will complete the data acquisition and interpretation to provide a fuller picture of this site. These results will be complemented by historical data obtained from the Cornwall Record Office on the type of extraction techniques employed at South Terras from ca. 1870–1930.
Acknowledgments
I thank Nuvia for the loan of the GroundhogTM system and Dr Black and Dr Almond for their help during the first field trip. I am grateful to the Boconnoc Estate for allowing me access to South Terras, and I thank Dr Imad Ahmed for inviting me to submit this article to the ECG Bulletin.
References for Paper 4
1. Smale, C. V. South Terras: Cornwall’s premier uranium and radium mine. Journal of the Royal Institution of Cornwall. New Series, 1993, 1(3), 304-321.
2. Dines, H. G. The metalliferous mining region of SW England. Volume 2. Memoirs of the Geological Survey of Great Britain (England and Wales), 1956, HMSO, London.
3. EDINA, South Terras. 2010, Digimap Collection.
4. Keith-Roach, M. J. The speciation, stability, solubility and biodegradation of organic co-contaminant radionuclide complexes: a review. Sci. Total Environ., 2008, 396, 1-1
5. Carlsen, L. The Role of Organics on the Migration of Radionuclides in the Geosphere: Final Report, Nuclear Science and Technology, Commission of the European Communities, Luxembourg, 1989.
6. Baker, A. C.; Toque, C. A review of the potential for radium from luminising activities to migrate in the environment. J. Radiol. Prot., 2005, 25, 127-140.
7. Moliner-Martinez, Y. Campíns-Falcó. P.; Worsfold, P. J.; Keith-Roach, M. J. The impact of a disused mine on uranium transport in the River Fal, South West England. J. Environ. Monit., 2004, 6, 907-913.

|

|
Views of the South Terras mining site, Cornwall. (Paper 4)
Paper 5 The Interest of Kinetic Considerations in Soil Heavy Metal Mobilization Assessment: An Overview
Nastaran Manouchehri, Stéphane Besançon, Alain Bermond
AgroParisTech, Analytical Chemistry Laboratory, 16, Rue Claude Bernard 750231 Paris Cedex 05
Introduction
Many agricultural and industrial activities such as sewage sludge, wastewaters, fertilizers, and smoke from factories result in trace metal pollution of soils. The deleterious effects of this contamination on the biota are of concern. However, it is known that the total concentration of a metal in soil is not a reliable indicator of its potential ecological risks. Current research in metal speciation in soil/solution systems attempts to determine the part of metal that is available to biota. Various single and sequential chemical extraction schemes have been devised to assess “reactive” or “labile” pools of metal in the soil solid phase (e.g. Barona and Romero, 1996; Tipping et al., 2003; Mocko and Waclawek, 2004; Feng et al., 2005; Young et al., 2006; Manouchehri and Bermond, 2009).
However, in most of these extraction methods, the metal concentration is measured at equilibrium whereas natural systems are generally subject to changing conditions and are not at equilibrium. For these dynamic systems, metal availability is thought to be controlled by kinetic factors (Errecalde et al., 1998; Ma et al., 1999; Fortin and Campbell, 2000; Hassler and Wilkinson, 2003; Slaveykova et al., 2003).
New techniques have been introduced to meet the challenge of understanding speciation in dynamic metal–soil/water systems. For example, kinetic fractionation (Bermond et al., 1998; Fangueiro et al., 2002; Bermond et al., 2005; Wasay et al., 2007) and Diffusive Gradients in Thin-films (DGT) (Zhang et al., 1995; Harper et al., 2000). Information on the dynamic behaviour of metals in the soil solid phase and their concentrations in pore solution is helpful for soil remediation procedures, the prediction of metal bioavailability, and risk assessment in general.
[In the accompanying web version of this paper, kinetic fractionation methods and developments in DGT are reviewed, and a soil/solution/resin batch model is used to illustrate kinetic monitoring.]
References for Paper 5
Barona, A.; Romero, R. F. (1996). Fractionation of lead in soils and its influence on the extractive cleaning with EDTA. Environ. Technol., 17, 63-70.
Bermond, A.; Ghestem, J.-P.; Yousfi, I. (1998). Kinetic approach to the chemical speciation of trace metals in soils. Analyst, 123, 785-790.
Bermond, A.; Varrault, G.; Sappin-Didier, V.; Mench, M. (2005). A kinetic approach to predict soil trace metal bioavailability: preliminary results. Plant Soil, 275, 21-29.
Errecalde, O.; Seidl, M.; Campbell, P. G. C. (1998). Influence of a low molecular weight metabolite (citrate) on the toxicity of cadmium and zinc to the unicellular green alga Selenastrum Capricornutum: An exception to the free-ion model. Water Res., 32, 419.
Fangueiro, D.; Bermond, A.; Santos, E.; Carapuça, H.; Duarte, A. (2002). Heavy metal mobility assessment in sediments based on a kinetic approach of the EDTA extraction : search for optimal experimental conditions. Anal. Chim. Acta, 459, 245-256.
Feng, M.-H.; Shan, X.-Q.; Zhang, S.; Wen, B. (2005). A comparison of the rhizosphere-based method with DTPA, EDTA, CaCl2 and NaNO3 extraction methods for prediction of bioavailability of metals in soil to barley. Environ. Pollut., 137, 231-240.
Fortin, C.; Campbell, P. G. C. (2000). Silver uptake by the green alga Chlamydomonas reinhardtii in relation to chemical speciation: influence of chloride. Environ. Toxicol. Chem., 19, 2769-2778.
Harper, M. P.; Davison, W.; Tych, W. (2000). DIFS-a Modelling and simulation tool for DGT induced trace metal remobilisation in sediments and soils. Environmental Modelling and Software, 15, 55-66.
Hassler, C. S.; Wilkinson, K. J. (2003). Failure of the biotic ligand and free-ion activity models to explain zinc bioaccumulation by Chlorella kesslerii. Environ. Toxicol. Chem., 22, 620-626.
Ma, H.; Kim, S. D.; Cha, D. K.; Allen, H. E. (1999). Effect of kinetics of complexation by humic acid on toxicity of copper to Ceriodaphina dubia. Environ. Toxicol. Chem., 18, 828-837.
Manouchehri, N.; Bermond, A. (2009). EDTA in soil science: A review of its application in soil trace metal investigations. Terr. Aquatic Environ. Toxicol., 3, 1-15.
Mocko, A.; Waclawek, W. (2004). Three-step extraction procedure for determination of heavy metals availability to vegetables. Anal. Bioanal. Chem., 380, 813-817.
Slaveykova, V. I.; Wilkinson, K. J.; Ceresa, A.; Pretsch, E. (2003). Role of fulvic acid on lead bioaccumulation by Chlorella kesslerii. Environ. Sci. Technol., 37, 1114-1121.
Tipping, E.; Rieuwerts, J. [and 6 co-authors] (2003). The solid-solution partitioning of heavy metals (Cu, Zn, Cd, Pb) in upland soils of England and Wales. Environ. Pollut., 125, 213-225.
Wasay, S. A.; Barrington, S. F.; Tokunagal, S.; Prasher, S. (2007). Kinetics of heavy metal desorption from three soils using citric acid, tartaric acid, and EDTA. J. Environ. Eng. Sci., 6, 611-622.
Young, S. D.; Zhang, H. [and 4 co-authors] (2006). Characterizing the availability of metals in contaminated soils. I. The solid phase: sequential extraction and isotopic dilution. Soil Use Manage., 21, 450-458.
Zhang, H.; Davison, W. (1995). Performance characteristics of diffusion gradients in thin films for the in situ measurement of trace metals in aqueous solution. Anal. Chem., 67, 3391-3400
Nastaran Manouchehri, Stéphane Besançon, Alain Bermond
AgroParisTech, Analytical Chemistry Laboratory, 16, Rue Claude Bernard 750231 Paris Cedex 05
Introduction
Many agricultural and industrial activities such as sewage sludge, wastewaters, fertilizers, and smoke from factories result in trace metal pollution of soils. The deleterious effects of this contamination on the biota are of concern. However, it is known that the total concentration of a metal in soil is not a reliable indicator of its potential ecological risks. Current research in metal speciation in soil/solution systems attempts to determine the part of metal that is available to biota. Various single and sequential chemical extraction schemes have been devised to assess “reactive” or “labile” pools of metal in the soil solid phase (e.g. Barona and Romero, 1996; Tipping et al., 2003; Mocko and Waclawek, 2004; Feng et al., 2005; Young et al., 2006; Manouchehri and Bermond, 2009).
However, in most of these extraction methods, the metal concentration is measured at equilibrium whereas natural systems are generally subject to changing conditions and are not at equilibrium. For these dynamic systems, metal availability is thought to be controlled by kinetic factors (Errecalde et al., 1998; Ma et al., 1999; Fortin and Campbell, 2000; Hassler and Wilkinson, 2003; Slaveykova et al., 2003).
New techniques have been introduced to meet the challenge of understanding speciation in dynamic metal–soil/water systems. For example, kinetic fractionation (Bermond et al., 1998; Fangueiro et al., 2002; Bermond et al., 2005; Wasay et al., 2007) and Diffusive Gradients in Thin-films (DGT) (Zhang et al., 1995; Harper et al., 2000). Information on the dynamic behaviour of metals in the soil solid phase and their concentrations in pore solution is helpful for soil remediation procedures, the prediction of metal bioavailability, and risk assessment in general.
[In the accompanying web version of this paper, kinetic fractionation methods and developments in DGT are reviewed, and a soil/solution/resin batch model is used to illustrate kinetic monitoring.]
References for Paper 5
Barona, A.; Romero, R. F. (1996). Fractionation of lead in soils and its influence on the extractive cleaning with EDTA. Environ. Technol., 17, 63-70.
Bermond, A.; Ghestem, J.-P.; Yousfi, I. (1998). Kinetic approach to the chemical speciation of trace metals in soils. Analyst, 123, 785-790.
Bermond, A.; Varrault, G.; Sappin-Didier, V.; Mench, M. (2005). A kinetic approach to predict soil trace metal bioavailability: preliminary results. Plant Soil, 275, 21-29.
Errecalde, O.; Seidl, M.; Campbell, P. G. C. (1998). Influence of a low molecular weight metabolite (citrate) on the toxicity of cadmium and zinc to the unicellular green alga Selenastrum Capricornutum: An exception to the free-ion model. Water Res., 32, 419.
Fangueiro, D.; Bermond, A.; Santos, E.; Carapuça, H.; Duarte, A. (2002). Heavy metal mobility assessment in sediments based on a kinetic approach of the EDTA extraction : search for optimal experimental conditions. Anal. Chim. Acta, 459, 245-256.
Feng, M.-H.; Shan, X.-Q.; Zhang, S.; Wen, B. (2005). A comparison of the rhizosphere-based method with DTPA, EDTA, CaCl2 and NaNO3 extraction methods for prediction of bioavailability of metals in soil to barley. Environ. Pollut., 137, 231-240.
Fortin, C.; Campbell, P. G. C. (2000). Silver uptake by the green alga Chlamydomonas reinhardtii in relation to chemical speciation: influence of chloride. Environ. Toxicol. Chem., 19, 2769-2778.
Harper, M. P.; Davison, W.; Tych, W. (2000). DIFS-a Modelling and simulation tool for DGT induced trace metal remobilisation in sediments and soils. Environmental Modelling and Software, 15, 55-66.
Hassler, C. S.; Wilkinson, K. J. (2003). Failure of the biotic ligand and free-ion activity models to explain zinc bioaccumulation by Chlorella kesslerii. Environ. Toxicol. Chem., 22, 620-626.
Ma, H.; Kim, S. D.; Cha, D. K.; Allen, H. E. (1999). Effect of kinetics of complexation by humic acid on toxicity of copper to Ceriodaphina dubia. Environ. Toxicol. Chem., 18, 828-837.
Manouchehri, N.; Bermond, A. (2009). EDTA in soil science: A review of its application in soil trace metal investigations. Terr. Aquatic Environ. Toxicol., 3, 1-15.
Mocko, A.; Waclawek, W. (2004). Three-step extraction procedure for determination of heavy metals availability to vegetables. Anal. Bioanal. Chem., 380, 813-817.
Slaveykova, V. I.; Wilkinson, K. J.; Ceresa, A.; Pretsch, E. (2003). Role of fulvic acid on lead bioaccumulation by Chlorella kesslerii. Environ. Sci. Technol., 37, 1114-1121.
Tipping, E.; Rieuwerts, J. [and 6 co-authors] (2003). The solid-solution partitioning of heavy metals (Cu, Zn, Cd, Pb) in upland soils of England and Wales. Environ. Pollut., 125, 213-225.
Wasay, S. A.; Barrington, S. F.; Tokunagal, S.; Prasher, S. (2007). Kinetics of heavy metal desorption from three soils using citric acid, tartaric acid, and EDTA. J. Environ. Eng. Sci., 6, 611-622.
Young, S. D.; Zhang, H. [and 4 co-authors] (2006). Characterizing the availability of metals in contaminated soils. I. The solid phase: sequential extraction and isotopic dilution. Soil Use Manage., 21, 450-458.
Zhang, H.; Davison, W. (1995). Performance characteristics of diffusion gradients in thin films for the in situ measurement of trace metals in aqueous solution. Anal. Chem., 67, 3391-3400
Paper 6 Speciation and distribution of arsenic for studies of availability and mobility in mining environments
Raquel Larios, Rodolfo Fernández-Martínez, M. Isabel Rucandio
Unidad de Espectroscopía, División de Química, Departamento de Tecnología, CIEMAT. Avda. Complutense, 22, Madrid, SPAIN.
Introduction
Arsenic is the twentieth most abundant element in the earth’s crust. It is a toxic trace element naturally present in all terrestrial and aquatic environments. High arsenic levels in water and soil endanger human health. Normal arsenic levels in soils range from 1 to 40 mg/kg [1], although some anthropogenic and volcanic activities can raise this upper limit. Various arsenic compounds are present in the environment and in biological systems [2]. Inorganic forms (arsenate and arsenite) are the predominant species, and at lower concentrations, monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA) are found.
Raquel Larios, Rodolfo Fernández-Martínez, M. Isabel Rucandio
Unidad de Espectroscopía, División de Química, Departamento de Tecnología, CIEMAT. Avda. Complutense, 22, Madrid, SPAIN.
Introduction
Arsenic is the twentieth most abundant element in the earth’s crust. It is a toxic trace element naturally present in all terrestrial and aquatic environments. High arsenic levels in water and soil endanger human health. Normal arsenic levels in soils range from 1 to 40 mg/kg [1], although some anthropogenic and volcanic activities can raise this upper limit. Various arsenic compounds are present in the environment and in biological systems [2]. Inorganic forms (arsenate and arsenite) are the predominant species, and at lower concentrations, monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA) are found.
Arsenic toxicity depends on its speciation. Trivalent arsenic compounds are considered to be more toxic than pentavalent arsenic compounds. An approximate order of decreasing toxicity is as follows: R3As > As(III) > (RAsO)n > As(V) > R2AsO(OH) > R4As+ > As(0); R = H > alkyl > aryl. Physico-chemical and environmental factors such as pH, redox potential, absorption-desorption processes and the presence of microorganisms contribute to arsenic speciation and toxicity.
The work described in this paper focuses on mercury mining areas in Asturias, a Spanish region with a historical tradition in mining and mineral processing. In particular, the extraction of mercury ores with a high arsenic content was quite widespread. As a result of these activities and related mechanical and chemical dispersion, significant amounts of arsenic have been released into the environment. Hyphenated analytical techniques are used to determine the extent and nature of arsenic pollution in the region.
Sampling area
The area we studied for this paper is located in Asturias (Northern Spain). Mieres and Pola de Lena are two districts where important mercury mines were exploited for decades and are nowadays abandoned. Three important mines belong to these areas: ”La Soterraña”, “La Peña-El Terronal” and “Los Rueldos” and were part of our study.
Sampling area
The area we studied for this paper is located in Asturias (Northern Spain). Mieres and Pola de Lena are two districts where important mercury mines were exploited for decades and are nowadays abandoned. Three important mines belong to these areas: ”La Soterraña”, “La Peña-El Terronal” and “Los Rueldos” and were part of our study.
|
Asturias was an important location for the production of mercury during the decade 1962-1972, with average annual productions of 15,000 flasks [3]. “La Peña-El Terronal” and “La Soterraña” were the most productive mines, located in Mieres and Pola de Lena districts, respectively, both in Central Asturias. The mines closed between 1973 and 1974 leaving a legacy of abandoned underground mines, extraction machinery and spoil heaps. In these Hg-mineralized areas, As is present in the form of As-rich pyrite, orpiment, realgar and occasionally arsenopyrite. Mine drainage and leachates from spoil heaps are often acidic with elevated As levels, leading to pollution of the Caudal River tributaries.
|

An abandoned mercury mine at Mieres, Asturias
|
Arsenic speciation in mining waters
Arsenic is a common constituent of many metal ores and a trace element in sulphides associated with waste deposits from mining activities. Both natural and accelerated weathering of these arsenic-bearing materials results in the release of arsenic into the waters. The concentration of As in unpolluted waters typically ranges 1-10 µg/L [4]. However, arsenic levels in water in exploited sulphide mineralization areas can be extremely high. In Asturias, arsenic concentrations up to 57,000 µg/L have been found [5].
Speciation analysis [6] indicates that inorganic arsenic species, As(III) and As(V), prevail in natural waters, especially in sulphide oxidation environments [7]. Methylated compounds, monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA) are not quantitatively important, and are only present in waters affected by industrial pollution [8].
Arsenic is a common constituent of many metal ores and a trace element in sulphides associated with waste deposits from mining activities. Both natural and accelerated weathering of these arsenic-bearing materials results in the release of arsenic into the waters. The concentration of As in unpolluted waters typically ranges 1-10 µg/L [4]. However, arsenic levels in water in exploited sulphide mineralization areas can be extremely high. In Asturias, arsenic concentrations up to 57,000 µg/L have been found [5].
Speciation analysis [6] indicates that inorganic arsenic species, As(III) and As(V), prevail in natural waters, especially in sulphide oxidation environments [7]. Methylated compounds, monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA) are not quantitatively important, and are only present in waters affected by industrial pollution [8].
Speciation of inorganic arsenic is a function of pH and redox potential (Figure 6). Under oxidising conditions at pH less than 6.9, H2AsO4− is dominant; at higher pH HAsO42− predominates. H3AsO4 and AsO43− are present in extremely acidic or alkaline conditions, respectively. Under reducing conditions at pH less than 9.2, the uncharged arsenic species H3AsO3 prevails. At moderate or high redox potentials, As may be present as pentavalent oxyanions (arsenate): H3AsO4, H2AsO4−, HAsO42−, and AsO43−. However, under more reducing conditions (acidic and mildly alkaline) and lower redox potential, trivalent arsenic species (H3AsO3) are found [9, 10].
|
In mining sites, acid mine drainage (AMD) is responsible for the release of relatively high levels of arsenic into surface and other waters. The high acidity generated during the oxidising processes gives rise to accelerated hydrolysis of minerals in the spoil materials, causing large quantities of the constituent elements to solubilise [11]. Arsenic minerals are associated with iron oxides or hydroxides, and a relationship between arsenic and iron speciation has been observed [11]. Correlations between the As(III)/(V) ratio and Fe(III) concentrations have been reported in waters from rivers affected by AMD. Also there is an inverse relationship between dissolved sulphate and arsenic which means that mobilisation of arsenic is not caused by oxidative dissolution of pyrite [12].
Analytical techniques for determining arsenic species |
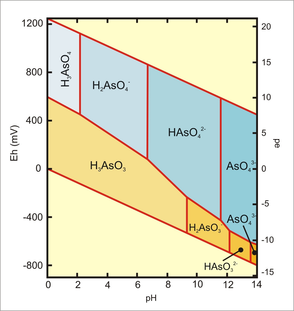
Figure 6 Eh-pH diagram for arsenic (III) and arsenic (V) oxyanions
|
High-performance liquid chromatography is widely used for the analysis of arsenic species. Buffers (mainly phosphate buffers) are usually employed as mobile phases at different pH values ranging from 4.5 to 8.5, and they can be used both in isocratic and in gradient modes [13]. For the separation of cationic species, cationic exchange columns are used e.g. Hamilton PRP X-200 [14] and Supelcosil LC- SCX [15]. For the determination of all species, two columns of different nature can be placed in parallel [16]. A favoured option for the separation of many arsenic compound is a Dionex Ion Pac As7 column [17]; separation of As(III), MMA, DMA, As(V), AsB, TMAO, AsC, TMA can be achieved within 14 minutes with nitric acid as a mobile phase in gradient mode.
For detection, Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) is commonly used, since it provides a great sensibility, multi-elemental capability, and large dynamic range. And coupling with HPLC is relatively easy, because the usual flow rates for HPLC are totally compatible with the uptake flow rate of an ICP system.
Hydride generation (HG), followed by spectrometry, is also popular. This technique is based on the formation of volatile hydrides by means of chemical treatment of a sample with a reducing agent, typically sodium borohydride. The advantage of this method is that the target arsenic species can be separated by volatilization from almost all other accompanying constituents in the sample through the HG process, so spectral and chemical interferences encountered in the detection systems are essentially eliminated. However, one drawback is that several organoarsenic compounds do not form volatile hydrides, so derivatisation methods to convert them into hydride-forming species are necessary. Microwave-assisted oxidation [18] and UV photo-oxidation [19] with potassium persulphate and sodium hydroxide have been used successfully for this task. Among the possible spectrometry techniques, atomic fluorescence (AFS) is the most attractive one, given its lowest detection limits.
For our field work in Asturias, HPLC-HG-AFS was the technique selected for the analysis of waters at the three mining sites. We showed that the four more common arsenic species could be separated using the technique (Figure 7), but in the actual samples from the Asturian mining sites, only inorganic species were found (Figure 8).
For detection, Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) is commonly used, since it provides a great sensibility, multi-elemental capability, and large dynamic range. And coupling with HPLC is relatively easy, because the usual flow rates for HPLC are totally compatible with the uptake flow rate of an ICP system.
Hydride generation (HG), followed by spectrometry, is also popular. This technique is based on the formation of volatile hydrides by means of chemical treatment of a sample with a reducing agent, typically sodium borohydride. The advantage of this method is that the target arsenic species can be separated by volatilization from almost all other accompanying constituents in the sample through the HG process, so spectral and chemical interferences encountered in the detection systems are essentially eliminated. However, one drawback is that several organoarsenic compounds do not form volatile hydrides, so derivatisation methods to convert them into hydride-forming species are necessary. Microwave-assisted oxidation [18] and UV photo-oxidation [19] with potassium persulphate and sodium hydroxide have been used successfully for this task. Among the possible spectrometry techniques, atomic fluorescence (AFS) is the most attractive one, given its lowest detection limits.
For our field work in Asturias, HPLC-HG-AFS was the technique selected for the analysis of waters at the three mining sites. We showed that the four more common arsenic species could be separated using the technique (Figure 7), but in the actual samples from the Asturian mining sites, only inorganic species were found (Figure 8).
|
As (V) was the only species found in all the samples from the three mining sites. This is agreement with the Eh-pH diagram, given the Eh and pH value of our samples (Soterraña: Eh~200mV, pH~8; La Peña-El Terronal: Eh~200mV, pH~8; Los Rueldos: upstream, before the mine: Eh~100mV, pH~7; downstream, after the mine: Eh~600, pH~3).
The most elevated arsenic concentrations occur at La Soterraña mine (~43,000 µg/L). Arsenic concentration is lower at La Peña-El Terronal (~1600 µg/L). This mine was subject to preventive measures in 2002 when spoil heap wastes from tailings were segregated in a secure landfill site to avoid the formation of leachates. The influence of AMD is evident in the Los Rueldos mining site. Samples collected upstream had low As concentrations. Here the waters are at neutral pH and oxidative capacity and heavy metal concentrations are low. Downstream from the spoil heap the aqueous environment is altered, solubilisation of heavy metals occurs and arsenic levels are raised (~10,000 µg/L). |
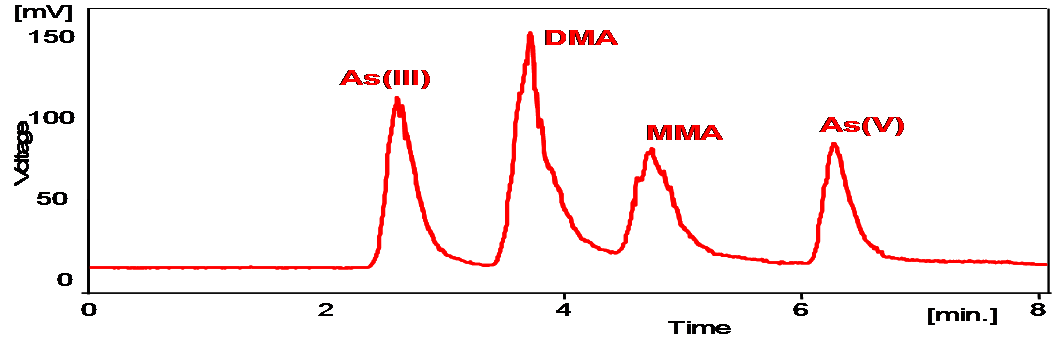
Figure 7: Chromatogram showing the separation and identification of four arsenic species using HPLC-HG-AFS.
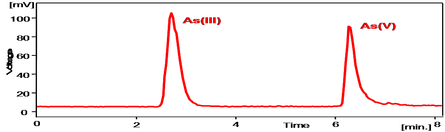
Figure 8: Chromatogram of the arsenic species found in Asturian mining samples using HPLC-HG-AFS
|
References for Paper 6
1. Ruiz-Chancho, M. J.; López-Sánchez, J. F.; Rubio, R. (2007). Analytical speciation as a tool to assess arsenic behaviour in soils polluted by mining. Anal. Bioanal. Chem., 387, 627-635.
2. Gong, Z.; Lu, X.; Ma, M.; Watt, C.; Le, X. C. (2002). Arsenic speciation analysis. Talanta, 58, 77-96.
3. SADEI, Datos y cifras de la economía asturiana; 1968-1991, Oviedo, Sociedad Asturiana de Estudios Económicos e Industriales, 1993, Oviedo, Spain.
4. Terlecka, E. (2005). Arsenic speciation analysis in water samples: A review of the hyphenated techniques. Environ. Monit. Assess., 107, 259-284.
5. Loredo, J.; Petit-Dominguez, M. D.; Ordóñez, A.; Galán, M. P.; Fernández-Martínez, R.; Alvárez, R.; Rucandio, M. I. (2010). Surface water monitoring in the mercury mining district of Asturias (Spain). J. Hazard. Mater., 176, 323-332.
6. Templeton, D. M.; Ariese, F.; Cornelis, R.; Danielsson, L. G.; Muntau, H.; Van Leeuwen, H. P.; Lobinski, R. (2000). Guidelines for terms related to chemical speciation and fractionation of elements. Definitions, structural aspects, and methodological approaches. Pure Appl. Chem., 72, 1453-1470.
7. Nordstrom, D. K.; Southam, G. (1997). Geomicrobiology of sulfide mineral oxidation. In: Banfield, J. F., Nealson, K.H. (eds.), Geomicrobiology: Interactions between Microbes and Minerals, Reviews in Mineralogy,Vol. 35. Mineralogical Society America, Washington, DC, pp. 361–390.
8. Smedley, P. L.; Kinniburgh, D. G. (2002). A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem., 17, 517–568.
9. Yan, X. P.; Kerrich, R.; Hendry, M. J. (2000). Distribution of arsenic(III), arsenic(V) and total inorganic arsenic in porewaters from a thick till and clay-rich aquitard sequence, Saskatchewan, Canada. Geochim. Cosmochim. Acta, 64, 2637–2648.
10. Mandal, B. K.; Kazno, T.; Suzuki, K. T. (2002). Arsenic round the world: A review. Talanta, 58, 201–235.
11. Sarmiento, A. M.; Nieto, J. M.; Casiot, C.; Elbaz-Poulichet, F.; Egal, M. (2009). Inorganic arsenic speciation at river basin scales: The Tinto and Odiel Rivers in the Iberian Pyrite Belt, SW Spain. Environ. Pollut., 157, 1202-1209.
12. Harvey, F. H.; Swartz, C. H.; Badruzzaman, A. B. M.; Keon-Blute, N.; Yu W.; Ashraf Ali, M.; Jay, J.; Beckie, R.; Niedan, V.; Brabander, D.; Oates, P. M.; Ashfaque, K. N.; Islam, S.; Hemond, H. F.; Ahmed, M. F. (2002). Arsenic mobility and groundwater extraction in Bangladesh. Science 298,1602-1606.
13. Koch, I.; Wang, C. A. L.; Ollson, C. A.; Cullen, W. R.; Reimer, K. J. (2000), The predominance of inorganic arsenic species in plants from Yellowknife, Northwest territories of Canada. Environ. Sci. Technol., 34, 22-26.
14. Súñer, M. A.; Devesa, V.; Rivas, I.; Velez, D.; Montoro, R. (2000). Speciation of cationic arsenic species in seafood by coupling liquid chromatography with hydride generation atomic fluorescence detection. J. Anal. At. Spectrom., 15, 1501-1507.
15. Feldmann, J.; John, K.; Pengrepecha, P. (2000). Arsenic metabolism in seaweed-eating sheep from Northern Scotland. Fresenius J. Anal. Chem., 368, 116-121.
16. Kuehnelt, D.; Goessler, W.; Irgolic, K. J. (1997). Arsenic compounds in terrestrial organisms: II. Arsenocholine in the mushroom amanita. Appl. Organomet. Chem., 11, 459-470.
17. Londensborough, S.; Mattusch, J.; Wennrich, R. (1999). Separation of organic and inorganic arsenic species by HPLC-ICPMS. Fresenius J. Anal. Chem., 363, 577-581.
18. Lamble, K. J.; Sperling, M.; Welz, B. (1996). Arsenic speciation in biological samples by on-line high performance liquid chromatography-microwave digestion hydride generation-atomic absorption spectrometry. Anal. Chim. Acta , 334, 261-270.
19. Zhang, X.; Cornelis, R.; de Kimpe, J.; Mees, L. (1996). Arsenic speciation in serum of uraemic patients based on liquid chromatography with hydride generation atomic absorption spectrometry and on-line UV photo-oxidation digestion. Anal. Chim. Acta, 319, 177-185.
1. Ruiz-Chancho, M. J.; López-Sánchez, J. F.; Rubio, R. (2007). Analytical speciation as a tool to assess arsenic behaviour in soils polluted by mining. Anal. Bioanal. Chem., 387, 627-635.
2. Gong, Z.; Lu, X.; Ma, M.; Watt, C.; Le, X. C. (2002). Arsenic speciation analysis. Talanta, 58, 77-96.
3. SADEI, Datos y cifras de la economía asturiana; 1968-1991, Oviedo, Sociedad Asturiana de Estudios Económicos e Industriales, 1993, Oviedo, Spain.
4. Terlecka, E. (2005). Arsenic speciation analysis in water samples: A review of the hyphenated techniques. Environ. Monit. Assess., 107, 259-284.
5. Loredo, J.; Petit-Dominguez, M. D.; Ordóñez, A.; Galán, M. P.; Fernández-Martínez, R.; Alvárez, R.; Rucandio, M. I. (2010). Surface water monitoring in the mercury mining district of Asturias (Spain). J. Hazard. Mater., 176, 323-332.
6. Templeton, D. M.; Ariese, F.; Cornelis, R.; Danielsson, L. G.; Muntau, H.; Van Leeuwen, H. P.; Lobinski, R. (2000). Guidelines for terms related to chemical speciation and fractionation of elements. Definitions, structural aspects, and methodological approaches. Pure Appl. Chem., 72, 1453-1470.
7. Nordstrom, D. K.; Southam, G. (1997). Geomicrobiology of sulfide mineral oxidation. In: Banfield, J. F., Nealson, K.H. (eds.), Geomicrobiology: Interactions between Microbes and Minerals, Reviews in Mineralogy,Vol. 35. Mineralogical Society America, Washington, DC, pp. 361–390.
8. Smedley, P. L.; Kinniburgh, D. G. (2002). A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem., 17, 517–568.
9. Yan, X. P.; Kerrich, R.; Hendry, M. J. (2000). Distribution of arsenic(III), arsenic(V) and total inorganic arsenic in porewaters from a thick till and clay-rich aquitard sequence, Saskatchewan, Canada. Geochim. Cosmochim. Acta, 64, 2637–2648.
10. Mandal, B. K.; Kazno, T.; Suzuki, K. T. (2002). Arsenic round the world: A review. Talanta, 58, 201–235.
11. Sarmiento, A. M.; Nieto, J. M.; Casiot, C.; Elbaz-Poulichet, F.; Egal, M. (2009). Inorganic arsenic speciation at river basin scales: The Tinto and Odiel Rivers in the Iberian Pyrite Belt, SW Spain. Environ. Pollut., 157, 1202-1209.
12. Harvey, F. H.; Swartz, C. H.; Badruzzaman, A. B. M.; Keon-Blute, N.; Yu W.; Ashraf Ali, M.; Jay, J.; Beckie, R.; Niedan, V.; Brabander, D.; Oates, P. M.; Ashfaque, K. N.; Islam, S.; Hemond, H. F.; Ahmed, M. F. (2002). Arsenic mobility and groundwater extraction in Bangladesh. Science 298,1602-1606.
13. Koch, I.; Wang, C. A. L.; Ollson, C. A.; Cullen, W. R.; Reimer, K. J. (2000), The predominance of inorganic arsenic species in plants from Yellowknife, Northwest territories of Canada. Environ. Sci. Technol., 34, 22-26.
14. Súñer, M. A.; Devesa, V.; Rivas, I.; Velez, D.; Montoro, R. (2000). Speciation of cationic arsenic species in seafood by coupling liquid chromatography with hydride generation atomic fluorescence detection. J. Anal. At. Spectrom., 15, 1501-1507.
15. Feldmann, J.; John, K.; Pengrepecha, P. (2000). Arsenic metabolism in seaweed-eating sheep from Northern Scotland. Fresenius J. Anal. Chem., 368, 116-121.
16. Kuehnelt, D.; Goessler, W.; Irgolic, K. J. (1997). Arsenic compounds in terrestrial organisms: II. Arsenocholine in the mushroom amanita. Appl. Organomet. Chem., 11, 459-470.
17. Londensborough, S.; Mattusch, J.; Wennrich, R. (1999). Separation of organic and inorganic arsenic species by HPLC-ICPMS. Fresenius J. Anal. Chem., 363, 577-581.
18. Lamble, K. J.; Sperling, M.; Welz, B. (1996). Arsenic speciation in biological samples by on-line high performance liquid chromatography-microwave digestion hydride generation-atomic absorption spectrometry. Anal. Chim. Acta , 334, 261-270.
19. Zhang, X.; Cornelis, R.; de Kimpe, J.; Mees, L. (1996). Arsenic speciation in serum of uraemic patients based on liquid chromatography with hydride generation atomic absorption spectrometry and on-line UV photo-oxidation digestion. Anal. Chim. Acta, 319, 177-185.